It’s not. This guidance is just one big “calm down people” letter from the agency. They publish these sorts of guidance every now and then because we as an industry can sometimes learn the wrong lessons.
So read the guidance, but don’t panic. You are either following it already or you just need to spend some time getting better at risk assessments and creating some matrix approaches.
In July 2022, the U.S. FDA issued a Warning Letterto the U.S. American company “Jost Chemical Co.” after having inspected its site in January 2022. The warning letter listedfour significant areas:
“Failure of your quality unit to ensure that quality-related complaints are investigated and resolved, and failure to extend investigations to other batches that may have been associated with a specific failure or deviation.”
“Failure to establish adequate written procedures for cleaning equipment and its release for use in manufacture of API.”
“Failure to ensure that all test procedures are scientifically sound and appropriate to ensure that your API conform to established standards of quality and purity, and failure to ensure laboratory data is complete and attributable.”
“Failure to exercise sufficient controls over computerized systems to prevent unauthorized access or changes to data, and failure to establish and follow written procedures for the operation and maintenance of your computerized systems.”
I offer them the above clip as a good mini-training. I recently watched the show, and my wife thought I was going to have several heart attacks.
In a serious nature, please do not short your efforts in data integrity.
In general, this report has few real planned actions and does not fill me with the hope of internal changes driving improvement.
One of the recommendations really stood out to me. Finding 2 states “Inadequate processes and lack of clarity related to whistleblower complaints may have delayed the FDA’s response to those complaints. A complaint sent via mail and other delivery systems by a confidential informant to agency leaders at FDA’s White Oak campus was not delivered to the addressees.”
Recommendation: The FDA should identify clear definitions for the terms “whistleblower,” “confidential informant,” and “informant,” and develop policies and provide training to staff regarding how to identify, escalate, and appropriately manage confidentiality of such complaints. The agency should also consider connecting complaints from such individuals to information received from product safety complaints, and product manufacturing concerns systems to support more complete access to all safety information. The FDA is evaluating how best to integrate this data to gain a holistic view of all FDA-regulated products and/or manufacturing facilities. The FDA should also review and update its mail and package delivery procedures to ensure that all mail and packages are delivered and received by addressees in a timely manner.
FDA Evaluation of Infant Formula Response
There is a real lack of whistleblower protection in this industry. Often when you hear about a crisis, from baby formula to Theranos to the opioid epidemic you have you have to ask “where were the good people at that company.” It can be rather disheartening. It has long been worrisome that the FDA does not have strong whistleblower protection in place, and to see how definitely that contributed to this debacle is just plain scary.
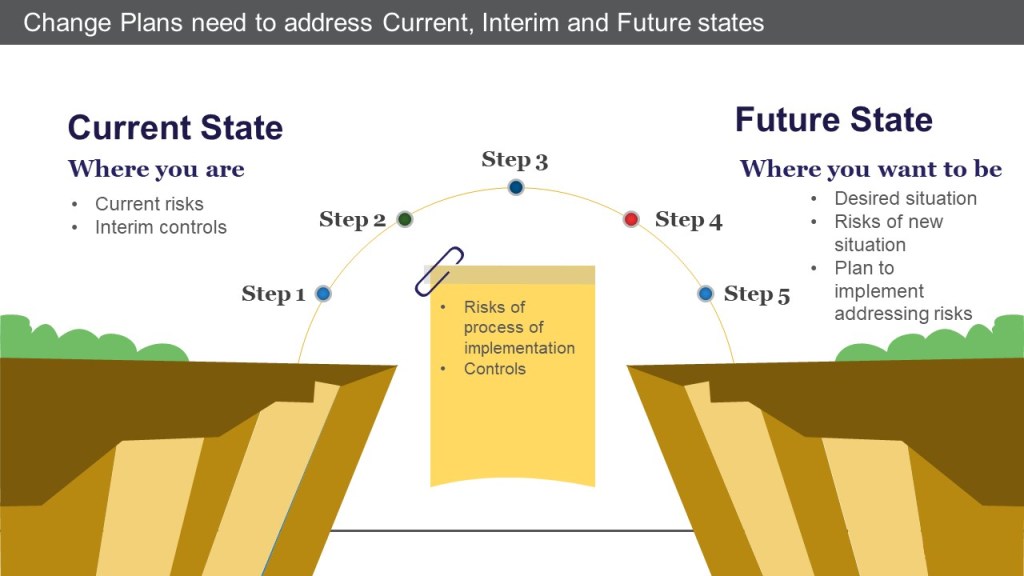
Potential risks with the current state (until changes are implemented) and any risks that might be temporarily introduced during the change process are adequately assessed.
Interim controls (short-term measures), as needed, are identified and implemented in a timely manner to monitor/mitigate risks associated with the current situation (until change implementation).
This section also recognizes that changes introduce an interim set of conditions that lead to implementation – from opening a machine through massive construction activities, and just about anything else.
A good change control does not just address what is necessary to implement the desired future state. It also contains interim controls for managing the current state until the future is implemented, and addresses all of the potential risks during the process of implementation.
Change plans address current, interim, and future states
Often the change control stems from an action plan in a CAPA record, where the interim controls are specifically called out and detailed. These may be adequate for a relatively brief period but are not realistically sustainable. Remember that these interim controls must also go through the appropriate change control process. Which may have been a separate change control or be part of this change control. In all cases, the change control should either determine that the CAPA plan is adequate or identify additional risks that were identified during change planning.
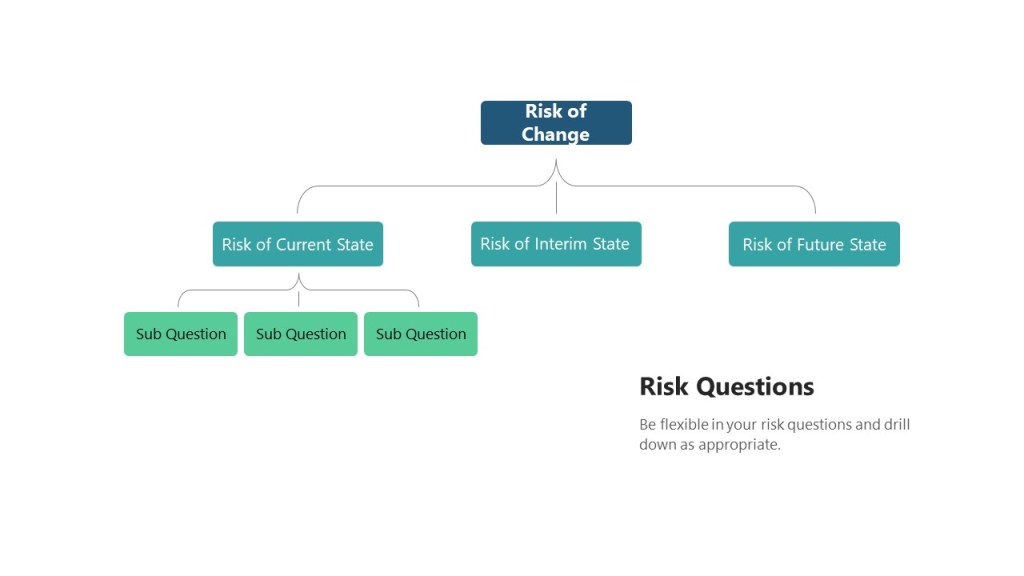
Risk assessments for change control should really have three basic risk questions:
What are the risks of the current state as we implement the change?
What are the risks of the implementation process?
What are the risks of the future state?
As discussed elsewhere, these questions are really branching trees.
The Risk Question branches in changes
Interim controls in a change are usually little temporary changes in the change plan that either mitigate occurrence or impact.
Interim Control Type
Mitigate Occurrence
Mitigate Impact
Examples
Every record processed will be reviewed for the event error before completion. Revised preventive maintenance procedure to require vibration test at next preventive maintenance. Increase cleaning frequency of incubator.
Add emergency response steps to address a valve that is malfunctioning. Add engineering check for pump prior to each use. Perform cleaning log review prior to use.
As temporary changes, it is important to determine how they will be implemented, if they require monitoring, and how they will go away. Often the process of implementation of the change removes the temporary changes, but that is not always the case.
Dear all, regarding question 20 “Is the effective date of the change (completion date) recorded and when appropriate the first batch manufactured recorded?” we are not sure how to handle it for multi-product changes. At our site we manufacture products of different customers. Sometimes we have changes, where f.e. documents of several products need to be adapted. How are the requirements defined in such cases? Do we need to report the first batch only once or for each product
At heart, this requirement is to help meet the regulatory requirement that “After implementation, an evaluation of the change should be undertaken to confirm the change objectives were achieved and that there was no deleterious impact on product quality.” (ICH Q9 part IV.B.3.d)
In order to perform an evaluation, you must be able to know when to start. This evaluation must be able to be performed on each and every product. It is from here we can settle on 1st batch. There are lots of ways to do this. The requirement is, for each and every product, to be able to state definitively when the change was first applied to the product.
Changes are monitored via ongoing monitoring systems to ensure maintenance of a state of control, and lessons learned are captured and shared/communicated. (Note: Activities such as Management Review, Annual Product Quality Review, Continuous Process Verification, Deviation Management and Complaint Monitoring can be useful in this regard.)
Having that definitive point allow all these activities to be effective.
The requirement is clear. We need to be able for any given change identify when it started impacting product.
All of the various Annual Product Review/Product Quality Review requirements also require the ability to evaluate all changes to the product in a given time frame.
Like many such requirements, there are multiple ways to do this. I think the requirement to capture first batch in a change control really stems from it, in many ways, being the easiest to do as you only have to manage it in the change control workflow. Though, in all honesty, it is also darn annoying with changes potentially being open more than a year to capture all possible products.
Based on our core requirement, there are several factors to consider:
There must be a way for a user to easily determine when a given change impacted a given product and to determine if a given batch was impacted by the changes
The user must be able to link any batch to all the changes that impacted it
The user must be able to run a report of all changes that impacted a product in a time frame
There are multiple ways of solving for this from the put a field in the change control to set of interfaces between the ERP and eQMS (and maybe MES).