As a leader, fostering critical thinking in my team and beyond is a core part of my job. Fostering critical thinking means an approach that encourages open-mindedness, curiosity, and structured problem-solving.
Encourage Questioning and Healthy Debate
It is essential to create an environment where team members feel comfortable questioning assumptions and engaging in constructive debates. Encourage them to ask “why” and explore different perspectives. This open dialogue promotes deeper thinking and prevents groupthink.
Foster a Culture of Curiosity
Inspire your team to ask questions and seek deeper understanding. Role model this behavior by starting meetings with thought-provoking “what if” scenarios or sharing your own curiosities. Celebrate curiosity and reward those who think outside the box.
Assign Stretch Assignments
Provide your team with challenging tasks that push them beyond their comfort zones. These stretch assignments force them to think critically, analyze information from multiple angles, and develop innovative solutions.
Promote Diverse Perspectives
Encourage diversity of thought within your team. Diverse backgrounds, experiences, and viewpoints can challenge assumptions and biases, leading to a more comprehensive understanding and better decision-making.
Engage in Collaborative Problem-Solving
Involve your team in decision-making processes and problem-solving exercises. Techniques like role reversal debates, where team members argue a point they disagree with, can help them understand different perspectives and refine their argumentative skills.
Provide Training and Resources
Offer training sessions on critical thinking techniques, such as SWOT analysis, root cause analysis, and logical fallacies. Equip your team with the tools and frameworks they need to think critically.
Lead by Example
As a leader, model critical thinking behaviors. Discuss your thought processes openly, question your assumptions, and show the value of critical evaluation in real-time decision-making. Your team will be more likely to emulate these habits.
Encourage Continuous Learning
Recommend learning resources, such as courses, articles, and books from diverse fields. Continuous learning can broaden perspectives and foster multifaceted thinking.
Embrace Feedback and Mistakes
Establish feedback loops within the team and create a safe environment where mistakes are treated as learning opportunities. Receiving and giving feedback helps refine understanding and overcome biases.
Implement Role-Playing Scenarios
Use role-playing scenarios to simulate real-world challenges. This helps team members practice critical thinking in a controlled environment, enhancing their ability to apply these skills in actual situations.
Build Into the Team Charter
Building these expectations into the team charter holds you and your team accountable.
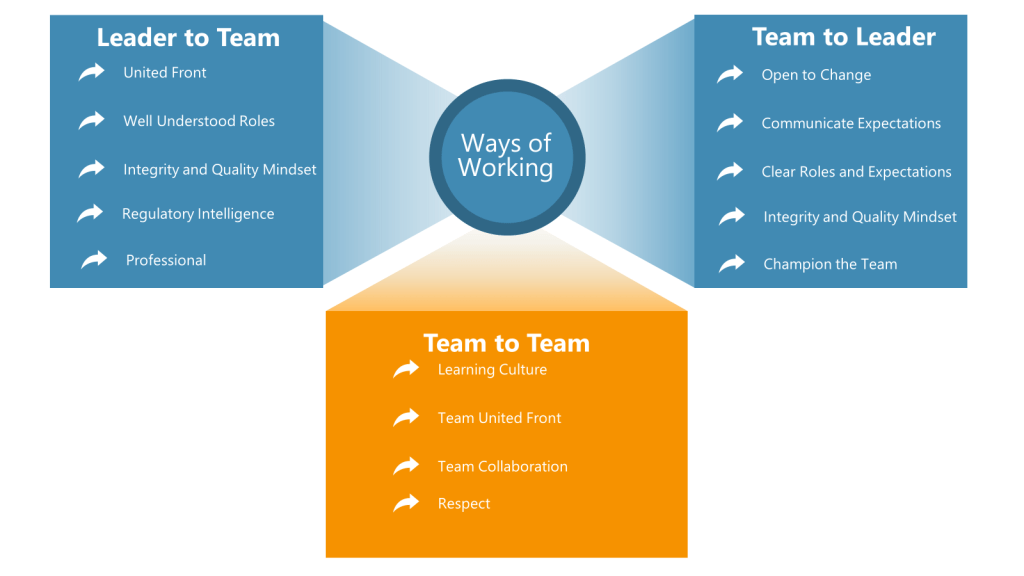
Value: Regulatory Intelligence
Definition: Stay current on industry regulations and guidances.
Desired Behaviors:
- I will dedicate time to reading industry-related guidance and regulation publications related to my job.
- I will share publications that I find interesting or applicable to my job with the team
- I will present to the team on at least one topic per year to share learnings with the team (or wider organization)
Value: Learning Culture
Definition: Share lessons learned from projects so the team can grow together and remain aligned. Engage in knowledge-sharing sessions.
Desired Behaviors:
- I will share lessons learned from each project with the wider team via the team channel and/or weekly team meeting.
- I will encourage team members to openly share their experiences, successes, and challenges without fear of judgement.
- I will update RAID log with decisions made by the team.
- I will identify possible process improvements and update the process improvement tracker.
Value: Team Collaboration
Definition: Willingness to help teammates when they reach out for input/help
Desired Behaviors:
- I will be supportive of my teammate’s requests for assistance
- I will engage and offer my SME advice when asked or help identify another SME to assist
- I will not ignore requests for input/help
- I will contribute to an environment where teammates can request help