I’m spending the month working at my company’s Toulouse site and my wife is with me and keeping a blog of her travel.
Author: Jeremiah Genest
Quality professional with over 20 years of experience. Gamer and storyteller with forty years of practice.
Industry 5.0, seriously?
This morning, an article landed in my inbox with the headline: “Why MES Remains the Digital Backbone, Even in Industry 5.0.” My immediate reaction? “You have got to be kidding me.” Honestly, that was also my second, third, and fourth reaction—each one a little more exasperated than the last. Sometimes, it feels like this relentless urge to slap a new number on every wave of technology is exactly why we can’t have nice things.
Curiosity got the better of me, though, and I clicked through. To my surprise, the article raised some interesting points. Still, I couldn’t help but wonder: do we really need another numbered revolution?
So, what exactly is Industry 5.0—and why is everyone talking about it? Let’s dig in.
The Origins and Evolution of Industry 5.0: From Japanese Society 5.0 to European Industrial Policy
The concept of Industry 5.0 emerged from a complex interplay of Japanese technological philosophy and European industrial policy, representing a fundamental shift from purely efficiency-driven manufacturing toward human-centric, sustainable, and resilient production systems. While the term “Industry 5.0” was formally coined by the European Commission in 2021, its intellectual foundations trace back to Japan’s Society 5.0 concept introduced in 2016, which envisioned a “super-smart society” that integrates cyberspace and physical space to address societal challenges. This evolution reflects a growing recognition that the Fourth Industrial Revolution’s focus on automation and digitalization, while transformative, required rebalancing to prioritize human welfare, environmental sustainability, and social resilience alongside technological advancement.
The Japanese Foundation: Society 5.0 as Intellectual Precursor
The conceptual roots of Industry 5.0 can be traced directly to Japan’s Society 5.0 initiative, which was first proposed in the Fifth Science and Technology Basic Plan adopted by the Japanese government in January 2016. This concept emerged from intensive deliberations by expert committees administered by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Ministry of Economy, Trade and Industry (METI) since 2014. Society 5.0 was conceived as Japan’s response to the challenges of an aging population, economic stagnation, and the need to compete in the digital economy while maintaining human-centered values.
The Japanese government positioned Society 5.0 as the fifth stage of human societal development, following the hunter-gatherer society (Society 1.0), agricultural society (Society 2.0), industrial society (Society 3.0), and information society (Society 4.0). This framework was designed to address Japan’s specific challenges, including rapid population aging, social polarization, and depopulation in rural areas. The concept gained significant momentum when it was formally presented by former Prime Minister Shinzo Abe in 2019 and received robust support from the Japan Business Federation (Keidanren), which saw it as a pathway to economic revitalization.
International Introduction and Recognition
The international introduction of Japan’s Society 5.0 concept occurred at the CeBIT 2017 trade fair in Hannover, Germany, where the Japanese Business Federation presented this vision of digitally transforming society as a whole. This presentation marked a crucial moment in the global diffusion of ideas that would later influence the development of Industry 5.0. The timing was significant, as it came just six years after Germany had introduced the Industry 4.0 concept at the same venue in 2011, creating a dialogue between different national approaches to industrial and societal transformation.
The Japanese approach differed fundamentally from the German Industry 4.0 model by emphasizing societal transformation beyond manufacturing efficiency. While Industry 4.0 focused primarily on smart factories and cyber-physical systems, Society 5.0 envisioned a comprehensive integration of digital technologies across all aspects of society to create what Keidanren later termed an “Imagination Society”. This broader vision included autonomous vehicles and drones serving depopulated areas, remote medical consultations, and flexible energy systems tailored to specific community needs.
European Formalization and Policy Development
The formal conceptualization of Industry 5.0 as a distinct industrial paradigm emerged from the European Commission’s research and innovation activities. In January 2021, the European Commission published a comprehensive 48-page white paper titled “Industry 5.0 – Towards a sustainable, human-centric and resilient European industry,” which officially coined the term and established its core principles. This document resulted from discussions held in two virtual workshops organized in July 2020, involving research and technology organizations and funding agencies across Europe.
The European Commission’s approach to Industry 5.0 represented a deliberate complement to, rather than replacement of, Industry 4.0. According to the Commission, Industry 5.0 “provides a vision of industry that aims beyond efficiency and productivity as the sole goals, and reinforces the role and the contribution of industry to society”. This formulation explicitly placed worker wellbeing at the center of production processes and emphasized using new technologies to provide prosperity beyond traditional economic metrics while respecting planetary boundaries.
Policy Integration and Strategic Objectives
The European conceptualization of Industry 5.0 was strategically aligned with three key Commission priorities: “An economy that works for people,” the “European Green Deal,” and “Europe fit for the digital age”. This integration demonstrates how Industry 5.0 emerged not merely as a technological concept but as a comprehensive policy framework addressing multiple societal challenges simultaneously. The approach emphasized adopting human-centric technologies, including artificial intelligence regulation, and focused on upskilling and reskilling European workers to prepare for industrial transformation.
The European Commission’s framework distinguished Industry 5.0 by its explicit focus on three core values: sustainability, human-centricity, and resilience. This represented a significant departure from Industry 4.0’s primary emphasis on efficiency and productivity, instead prioritizing environmental responsibility, worker welfare, and system robustness against external shocks such as the COVID-19 pandemic. The Commission argued that this approach would enable European industry to play an active role in addressing climate change, resource preservation, and social stability challenges.
Conceptual Evolution and Theoretical Development
From Automation to Human-Machine Collaboration
The evolution from Industry 4.0 to Industry 5.0 reflects a fundamental shift in thinking about the role of humans in automated production systems. While Industry 4.0 emphasized machine-to-machine communication, Internet of Things connectivity, and autonomous decision-making systems, Industry 5.0 reintroduced human creativity and collaboration as central elements. This shift emerged from practical experiences with Industry 4.0 implementation, which revealed limitations in purely automated approaches and highlighted the continued importance of human insight, creativity, and adaptability.
Industry 5.0 proponents argue that the concept represents an evolution rather than a revolution, building upon Industry 4.0’s technological foundation while addressing its human and environmental limitations. The focus shifted toward collaborative robots (cobots) that work alongside human operators, combining the precision and consistency of machines with human creativity and problem-solving capabilities. This approach recognizes that while automation can handle routine and predictable tasks effectively, complex problem-solving, innovation, and adaptation to unexpected situations remain distinctly human strengths.
Academic and Industry Perspectives
The academic and industry discourse around Industry 5.0 has emphasized its role as a corrective to what some viewed as Industry 4.0’s overly technology-centric approach. Scholars and practitioners have noted that Industry 4.0’s focus on digitalization and automation, while achieving significant efficiency gains, sometimes neglected human factors and societal impacts. Industry 5.0 emerged as a response to these concerns, advocating for a more balanced approach that leverages technology to enhance rather than replace human capabilities.
The concept has gained traction across various industries as organizations recognize the value of combining technological sophistication with human insight. This includes applications in personalized manufacturing, where human creativity guides AI systems to produce customized products, and in maintenance operations, where human expertise interprerets data analytics to make complex decisions about equipment management416. The approach acknowledges that successful industrial transformation requires not just technological advancement but also social acceptance and worker engagement.
Timeline and Key Milestones
The development of Industry 5.0 can be traced through several key phases, beginning with Japan’s internal policy deliberations from 2014 to 2016, followed by international exposure in 2017, and culminating in European formalization in 2021. The COVID-19 pandemic played a catalytic role in accelerating interest in Industry 5.0 principles, as organizations worldwide experienced the importance of resilience, human adaptability, and sustainable practices in maintaining operations during crisis conditions.
The period from 2017 to 2020 saw growing academic and industry discussion about the limitations of purely automated approaches and the need for more human-centric industrial models. This discourse was influenced by practical experiences with Industry 4.0 implementation, which revealed challenges in areas such as worker displacement, skill gaps, and environmental sustainability. The European Commission’s workshops in 2020 provided a formal venue for consolidating these concerns into a coherent policy framework.
Contemporary Developments and Future Trajectory
Since the European Commission’s formal introduction of Industry 5.0 in 2021, the concept has gained international recognition and adoption across various sectors. The approach has been particularly influential in discussions about sustainable manufacturing, worker welfare, and industrial resilience in the post-pandemic era. Organizations worldwide are beginning to implement Industry 5.0 principles, focusing on human-machine collaboration, environmental responsibility, and system robustness.
The concept continues to evolve as practitioners gain experience with its implementation and as new technologies enable more sophisticated forms of human-machine collaboration. Recent developments have emphasized the integration of artificial intelligence with human expertise, the application of circular economy principles in manufacturing, and the development of resilient supply chains capable of adapting to global disruptions. These developments suggest that Industry 5.0 will continue to influence industrial policy and practice as organizations seek to balance technological advancement with human and environmental considerations.
Evaluating Industry 5.0 Concepts
While I am naturally suspicious of version numbers on frameworks, and certainly exhausted by the Industry 4.0/Quality 4.0 advocates, the more I read about industry 5.0 the more the core concepts resonated with me. Industry 5.0 challenges manufacturers to reshape how they think about quality, people, and technology. And this resonates on what has always been the fundamental focus of this blog: robust Quality Units, data integrity, change control, and the organizational structures needed for true quality oversight.
Human-Centricity: From Oversight to Empowerment
Industry 5.0’s defining feature is its human-centric approach, aiming to put people back at the heart of manufacturing. This aligns closely with my focus on decision-making, oversight, and continuous improvement.
Collaboration Between Humans and Technology
I frequently address the pitfalls of siloed teams and the dangers of relying solely on either manual or automated systems for quality management. Industry 5.0’s vision of human-machine collaboration—where AI and automation support, but don’t replace, expert judgment—mirrors this blog’s call for integrated quality systems.
Proactive, Data-Driven Quality
To say that a central theme in my career has been how reactive, paper-based, or poorly integrated systems lead to data integrity issues and regulatory citations would be an understatement. Thus, I am fully aligned with the advocacy for proactive, real-time management utilizing AI, IoT, and advanced analytics. This continued shift from after-the-fact remediation to predictive, preventive action directly addresses the recurring compliance gaps we continue to struggle with. This blog’s focus on robust documentation, risk-based change control, and comprehensive batch review finds a natural ally in Industry 5.0’s data-driven, risk-based quality management systems.
Sustainability and Quality Culture
Another theme on this blog is the importance of management support and a culture of quality—elements that Industry 5.0 elevates by integrating sustainability and social responsibility into the definition of quality itself. Industry 5.0 is not just about defect prevention; it’s about minimizing waste, ensuring ethical sourcing, and considering the broader impact of manufacturing on people and the planet. This holistic view expands the blog’s advocacy for independent, well-resourced Quality Units to include environmental and social governance as core responsibilities. Something I perhaps do not center as much in my practice as I should.
Democratic Leadership
The principles of democratic leadership explored extensively on this blog provide a critical foundation for realizing the human-centric aspirations of Industry 5.0. Central to the my philosophy is decentralizing decision-making and fostering psychological safety—concepts that align directly with Industry 5.0’s emphasis on empowering workers through collaborative human-machine ecosystems. By advocating for leadership models that distribute authority to frontline employees and prioritize transparency, this blog’s framework mirrors Industry 5.0’s rejection of rigid hierarchies in favor of agile, worker-driven innovation. The emphasis on equanimity—maintaining composed, data-driven responses to quality challenges—resonates with Industry 5.0’s vision of resilient systems where human judgment guides AI and automation. This synergy is particularly evident in the my analysis of decentralized decision-making, which argues that empowering those closest to operational realities accelerates problem-solving while building ownership—a necessity for Industry 5.0’s adaptive production environments. The European Commission’s Industry 5.0 white paper explicitly calls for this shift from “shareholder to stakeholder value,” a transition achievable only through the democratic leadership practices championed in the blog’s critique of Taylorist management models. By merging technological advancement with human-centric governance, this blog’s advocacy for flattened hierarchies and worker agency provides a blueprint for implementing Industry 5.0’s ideals without sacrificing operational rigor.
Convergence and Opportunity
While I have more than a hint of skepticism about the term Industry 5.0, I acknowledge its reliance on the foundational principles that I consider crucial to quality management. By integrating robust organizational quality structures, empowered individuals, and advanced technology, manufacturers can transcend mere compliance to deliver sustainable, high-quality products in a rapidly evolving world. For quality professionals, the implication is clear: the future is not solely about increased automation or stricter oversight but about more intelligent, collaborative, and, importantly, human-centric quality management. This message resonates deeply with me, and it should with you as well, as it underscores the value and importance of our human contribution in this process.
Key Sources on Industry 5.0
Here is a curated list of foundational and authoritative sources for understanding Industry 5.0, including official reports, academic articles, and expert analyses that I found most helpful when evaluating the concept of Industry 5.0:
- European Commission, Directorate-General for Research and Innovation: “Industry 5.0: Towards a sustainable, human-centric and resilient European industry” (2021)
- The seminal report that formally introduces and defines Industry 5.0, outlining its pillars and societal vision.
- TWI Global: “What is Industry 5.0? (Top 5 Things You Need To Know)“
- An accessible overview of Industry 5.0, its technologies, and its human-centric approach.
- Automation.com: “The Three Pillars of Industry 5.0“
- Explains the central pillars—human focus, sustainability, and resiliency—and their practical implications for organizations.
- Forbes: “What Is Industry 5.0 And How It Will Radically Change Your Business Strategy?” by Jeroen Kraaijenbrink
- Discusses the shift from economic to societal value and the broader strategic implications of Industry 5.0.
- ScienceDirect: “Industry 5.0: A new strategy framework for sustainability“
- Presents a novel framework for manufacturing strategy in the Industry 5.0 paradigm, with academic rigor and case studies.
- ScienceDirect: “Industry 5.0 – Past, Present, and Near Future“
- A scholarly review of the evolution, current research trends, and future directions of Industry 5.0, with links to further academic literature.
- Grand View Research: “Industry 5.0 Market Size & Share | Industry Report, 2030“
- Provides market analysis, trends, and forecasts related to Industry 5.0 technologies and adoption.
- UNESCO: “Japan pushing ahead with Society 5.0 to overcome chronic social challenges”
- Explores the Japanese Society 5.0 initiative, which heavily influenced the development of Industry 5.0 concepts.
- Eviden: “How Industry 5.0 puts values first“
- Focuses on the value-driven evolution of Industry 5.0, highlighting its impact on people, sustainability, and resilience.
- Proaction International Blog: “Industry 5.0: Revolutionizing Work by Putting People First“
- Explores the people-centric transformation of work and management under Industry 5.0.
Quality Unit Oversight Failures: A Critical Analysis of Recent FDA Warning Letters
The continued trend in FDA warning letters citing Quality Unit (QU) deficiencies highlights a concerning reality across pharmaceutical manufacturing operations worldwide. Three warning letters recently issued to pharmaceutical companies in China, India, and Malaysia reveal fundamental weaknesses in Quality Unit oversight that extend beyond isolated procedural failures to indicate systemic quality management deficiencies. These regulatory actions demonstrate the FDA’s continued emphasis on the Quality Unit as the cornerstone of pharmaceutical quality systems, with expectations that these units function as independent guardians of product quality with sufficient authority, resources, and expertise. This analysis examines the specific deficiencies identified across recent warning letters, identifies patterns of Quality Unit organizational failures, explores regulatory expectations, and provides strategic guidance for building robust quality oversight capabilities that meet evolving compliance standards.
Recent FDA Warning Letters Highlighting Critical Quality Unit Deficiencies
Multiple Geographic Regions Under Scrutiny
The FDA has continues to provide an intense focus on Quality Unit oversight through a series of warning letters targeting pharmaceutical operations across Asia. As highlighted in a May 19, 2025 GMP Compliance article, three notable warning letters targeted specific Quality Unit failures across multiple regions. The Chinese manufacturer failed to establish an adequate Quality Unit with proper authority to oversee manufacturing operations, particularly in implementing change control procedures and conducting required periodic product reviews. Similarly, the Indian manufacturer’s Quality Unit failed to implement controls ensuring data integrity, resulting in unacceptable documentation practices including torn batch records, damaged testing chromatograms, and improperly completed forms. The Malaysian facility, producing OTC products, showed failures in establishing adequate training programs and performing appropriate product reviews, further demonstrating systemic quality oversight weaknesses. These geographically diverse cases indicate that Quality Unit deficiencies represent a global challenge rather than isolated regional issues.
Historical Context of Regulatory Concerns
FDA’s focus on Quality Unit responsibilities isn’t new. A warning letter to a Thai pharmaceutical company earlier in 2024 cited Quality Unit deficiencies including lack of control over manufacturing operations, inadequate documentation of laboratory preparation, and insufficient review of raw analytical data. These issues allowed concerning practices such as production staff altering master batch records and using erasable markers on laminated sheets for production records. Another notable case involved Henan Kangdi Medical Devices, where in January 2020 the FDA stated explicitly that “significant findings in this letter indicate that your quality unit is not fully exercising its authority and/or responsibilities”. The consistent regulatory focus across multiple years suggests pharmaceutical manufacturers continue to struggle with properly empowering and positioning Quality Units within their organizational structures.
Geographic Analysis of Quality Unit Failures: Emerging vs. Mature Regulatory Markets
These FDA warning letters highlighting Quality Unit (QU) deficiencies reveal significant disparities between pharmaceutical manufacturing practices in emerging markets (e.g., China, India, Malaysia, Thailand) and mature regulatory jurisdictions (e.g., the U.S., EU, Japan). These geographic differences reflect systemic challenges tied to regulatory infrastructure, economic priorities, and technological adoption.
In emerging markets, structural weaknesses in regulatory oversight and quality culture dominate QU failures. For example, Chinese manufacturers like Linghai ZhanWang Biotechnology (2025) and Henan Kangdi (2019) faced FDA action because their Quality Units lacked the authority to enforce CGMP standards, with production teams frequently overriding quality decisions. Similarly, Indian facilities cited in 2025 warnings struggled with basic data integrity controls, including torn paper records and unreviewed raw data—issues exacerbated by domestic regulatory bodies like India’s CDSCO, which inspects fewer than 2% of facilities annually. These regions often prioritize production quotas over compliance, leading to under-resourced Quality Units and inadequate training programs, as seen in a 2025 warning letter to a Malaysian OTC manufacturer whose QU staff lacked GMP training. Supply chain fragmentation further complicates oversight, particularly in contract manufacturing hubs like Thailand, where a 2024 warning letter noted no QU review of outsourced laboratory testing.
By contrast, mature markets face more nuanced QU challenges tied to technological complexity and evolving regulatory expectations. In the U.S. and EU, recent warnings highlight gaps in Quality Units’ understanding of advanced manufacturing technologies, such as continuous manufacturing processes or AI-driven analytics. A 2024 EU warning letter to a German API manufacturer, for instance, cited cybersecurity vulnerabilities in electronic batch records—a stark contrast to emerging markets’ struggles with paper-based systems. While data integrity remains a global concern, mature markets grapple with sophisticated gaps like inadequate audit trails in cloud-based laboratory systems, whereas emerging economies face foundational issues like erased entries or unreviewed chromatograms. Regulatory scrutiny also differs: FDA inspection data from 2023 shows QU-related citations in just 6.2% of U.S. facilities versus 23.1% in Asian operations, reflecting stronger baseline compliance in mature jurisdictions.
Case comparisons illustrate these divergences. At an Indian facility warned in 2025, production staff routinely overruled QU decisions to meet output targets, while a 2024 U.S. warning letter described a Quality Unit delaying batch releases due to inadequate validation of a new AI-powered inventory system. Training gaps also differ qualitatively: emerging-market QUs often lack basic GMP knowledge, whereas mature-market teams may struggle with advanced tools like machine learning algorithms.
These geographic trends have strategic implications. Emerging markets require foundational investments in QU independence, such as direct reporting lines to executive leadership, and adoption of centralized digital systems to mitigate paper-record risks. Partnerships with mature-market firms could accelerate quality culture development. Meanwhile, mature jurisdictions must modernize QU training programs to address rapidly changing technologies and strengthen oversight of decentralized production models.
Data Integrity as a Critical Quality Unit Responsibility
Data integrity issues feature prominently in recent enforcement actions, reflecting the Quality Unit’s crucial role as guardian of trustworthy information. The FDA frequently requires manufacturers with data integrity deficiencies to engage third-party consultants to conduct comprehensive investigations into record inaccuracies across all laboratories, manufacturing operations, and relevant systems. These remediation efforts must identify numerous potential issues including omissions, alterations, deletions, record destruction, non-contemporaneous record completion, and other deficiencies that undermine data reliability. Thorough risk assessments must evaluate potential impacts on product quality, with companies required to implement both interim protective measures and comprehensive long-term corrective actions. These requirements underscore the fundamental importance of the Quality Unit in ensuring that product decisions are based on accurate, complete, and trustworthy data.
Patterns of Quality Unit Organizational Failures
Insufficient Authority and Resources
A recurring theme across warning letters is Quality Units lacking adequate authority or resources to fulfill their responsibilities effectively. The FDA’s warning letter to Linghai ZhanWang Biotechnology Co. in February 2025 cited violations that demonstrated the company’s Quality Unit couldn’t effectively ensure compliance with CGMP regulations. Similarly, Lex Inc. faced regulatory action when its “quality system was inadequate” because the Quality Unit “did not provide adequate oversight for the manufacture of over-the-counter (OTC) drug products”.
These cases reflect a fundamental organizational failure to empower Quality Units with sufficient authority and resources to perform their essential functions. Without proper positioning within the organizational hierarchy, Quality Units cannot effectively challenge manufacturing decisions that might compromise product quality or regulatory compliance, creating systemic vulnerabilities.
Documentation and Data Management Deficiencies
Quality Units frequently demonstrate inadequate oversight of documentation and data management processes, allowing significant compliance risks to emerge. According to FDA warning letters, these issues include torn batch records, incompletely documented laboratory preparation, inadequate retention of weight printouts, and insufficient review of raw analytical data. One particularly concerning practice involved “production records on laminated sheets using erasable markers that could be easily altered or lost,” representing a fundamental breakdown of documentation control. These examples demonstrate how Quality Unit failures in documentation oversight directly enable data integrity issues that can undermine the reliability of manufacturing records, ultimately calling product quality into question. Effective Quality Units must establish robust systems for ensuring complete, accurate, and contemporaneous documentation throughout the manufacturing process.
Inadequate Change Control and Risk Assessment
Change control deficiencies represent another significant pattern in Quality Unit failures. Warning letters frequently cite the Quality Unit’s failure to ensure appropriate change control procedures, highlighting inadequate risk assessments as a particular area of concern. FDA inspectors have found that inadequate change control practices present significant compliance risks, with change control appearing among the top ten FDA 483 violations. These deficiencies often involve failure to evaluate the potential impact of changes on product quality, incomplete documentation of changes, and improper execution of change implementation. Effective Quality Units must establish robust change control processes that include thorough risk assessments, appropriate approvals, and verification that changes have not adversely affected product quality.
Insufficient Batch Release and Production Record Review
Quality Units regularly fail to conduct adequate reviews of production records and properly execute batch release procedures. A frequent citation in warning letters involves the Quality Unit’s failure to “review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated”. In several cases, the Quality Unit reviewed only analytical results entered into enterprise systems without examining the underlying raw analytical data, creating significant blind spots in quality oversight. This pattern demonstrates a superficial approach to batch review and release decisions that fails to fulfill the Quality Unit’s fundamental responsibility to ensure each batch meets all established specifications before distribution. Comprehensive batch record review is essential for detecting anomalies that might indicate quality or compliance issues requiring investigation.
Regulatory Expectations for Effective Quality Units
Core Quality Unit Responsibilities
The FDA has clearly defined the essential responsibilities of the Quality Unit through regulations, guidance documents, and enforcement actions. According to 21 CFR 211.22, the Quality Unit must “have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products”. Additionally, the unit must “review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated”. FDA guidance elaborates that the Quality Unit’s duties include “ensuring that controls are implemented and completed satisfactorily during manufacturing operations” and “ensuring that developed procedures and specifications are appropriate and followed”. These expectations establish the Quality Unit as both guardian and arbiter of quality throughout the manufacturing process, with authority to make critical decisions regarding product acceptability.
Independence and Organizational Structure
Regulatory authorities expect Quality Units to maintain appropriate independence from production units to prevent conflicts of interest. FDA guidance specifically states that “under a quality system, it is normally expected that the product and process development units, the manufacturing units, and the QU will remain independent”. This separation ensures that quality decisions remain objective and focused on product quality rather than production metrics or efficiency considerations. While the FDA acknowledges that “in very limited circumstances, a single individual can perform both production and quality functions,” such arrangements require additional safeguards including “another qualified individual, not involved in the production operation, conduct[ing] an additional, periodic review of QU activities”. This guidance underscores the critical importance of maintaining appropriate separation between quality and production responsibilities.
Quality System Integration
Regulatory authorities increasingly view the Quality Unit as the central coordinator of a comprehensive quality system. The FDA’s guidance document “Quality Systems Approach to Pharmaceutical CGMP Regulations” positions the Quality Unit as responsible for creating, monitoring, and implementing the entire quality system. This expanded view recognizes that while the Quality Unit doesn’t assume responsibilities belonging to other organizational units, it plays a crucial role in ensuring that all departments understand and fulfill their quality-related responsibilities. The Quality Unit must therefore establish appropriate communication channels and collaborative mechanisms with other functional areas while maintaining the independence necessary to make objective quality decisions. This integrated approach recognizes that quality management extends beyond a single department to encompass all activities affecting product quality.
Strategic Approaches to Strengthening Quality Unit Effectiveness
Comprehensive Quality System Assessment
Organizations facing Quality Unit deficiencies should begin remediation with a thorough assessment of their entire pharmaceutical quality system. Warning letters frequently require companies to conduct “a comprehensive assessment and remediation plan to ensure your QU is given the authority and resources to effectively function”. This assessment should examine whether procedures are “robust and appropriate,” how the Quality Unit provides oversight “throughout operations to evaluate adherence to appropriate practices,” the effectiveness of batch review processes, and the Quality Unit’s investigational capabilities. A thorough gap analysis should compare current practices against regulatory requirements and industry best practices to identify specific areas requiring improvement. This comprehensive assessment provides the foundation for developing targeted remediation strategies that address the root causes of Quality Unit deficiencies.
Establishing Clear Roles and Adequate Resources
Effective remediation requires clearly defining Quality Unit roles and ensuring adequate resources to fulfill regulatory responsibilities. FDA warning letters frequently cite the absence of “written procedures for QU roles and responsibilities” as a significant deficiency. Organizations must develop detailed written procedures that clearly articulate the Quality Unit’s authority and responsibilities, including approval or rejection authority for components and drug products, review of production records, and oversight of quality-impacting procedures and specifications. Additionally, companies must assess whether Quality Units have sufficient staffing with appropriate qualifications and training to effectively execute these responsibilities. This assessment should consider both the number of personnel and their technical capabilities relative to the complexity of manufacturing operations and product portfolio.
Implementing Robust Data Integrity Controls
Data integrity represents a critical area requiring focused attention from Quality Units. Companies must implement comprehensive data governance systems that ensure records are attributable, legible, contemporaneous, original, and accurate (ALCOA principles). Quality Units should establish oversight mechanisms for all quality-critical data, including laboratory results, manufacturing records, and investigation documentation. These systems must include appropriate controls for paper records and electronic data, with verification processes to ensure consistency between different data sources. Quality Units should also implement risk-based audit programs that regularly evaluate data integrity practices across all manufacturing and laboratory operations. These controls provide the foundation for trustworthy data that supports sound quality decisions and regulatory compliance.
Developing Management Support and Quality Culture
Sustainable improvements in Quality Unit effectiveness require strong management support and a positive quality culture throughout the organization. FDA warning letters specifically call for “demonstration of top management support for quality assurance and reliable operations, including timely provision of resources to address emerging manufacturing and quality issues”. Executive leadership must visibly champion quality as an organizational priority and empower the Quality Unit with appropriate authority to fulfill its responsibilities effectively. Organizations should implement programs that promote quality awareness at all levels, with particular emphasis on the shared responsibility for quality across all departments. Performance metrics and incentive structures should align with quality objectives to reinforce desired behaviors and decision-making patterns. This culture change requires consistent messaging, appropriate resource allocation, and leadership accountability for quality outcomes.
Conclusion
FDA warning letters reveal persistent Quality Unit deficiencies across global pharmaceutical operations, with significant implications for product quality and regulatory compliance. The patterns identified—including insufficient authority and resources, documentation and data management weaknesses, inadequate change control, and ineffective batch review processes—highlight the need for fundamental improvements in how Quality Units are structured, resourced, and empowered within pharmaceutical organizations. Regulatory expectations clearly position the Quality Unit as the cornerstone of effective pharmaceutical quality systems, with responsibility for ensuring that all operations meet established quality standards through appropriate oversight, review, and decision-making processes.
Addressing these challenges requires a strategic approach that begins with comprehensive assessment of current practices, establishment of clear roles and responsibilities, implementation of robust data governance systems, and development of a supportive quality culture. Organizations that successfully strengthen their Quality Units can not only avoid regulatory action but also realize significant operational benefits through more consistent product quality, reduced manufacturing deviations, and more efficient operations. As regulatory scrutiny of Quality Unit effectiveness continues to intensify, pharmaceutical manufacturers must prioritize these improvements to ensure sustainable compliance and protect patient safety in an increasingly complex manufacturing environment.
Key Warning Letters Discussed
- Linghai ZhanWang Biotechnology Co., Ltd. (China) — February 25, 2025
- (For the original FDA letter, search the FDA Warning Letters database for “Linghai ZhanWang Biotechnology Co” and the date “02/25/2025”)
- Henan Kangdi Medical Devices Co. Ltd. (China) — December 3, 2019
- (For the original FDA letter, search the FDA Warning Letters database for “Henan Kangdi Medical Devices” and the date “12/03/2019”)
- Drug Manufacturing Facility in Thailand — February 27, 2024
- (For the original FDA letter, search the FDA Warning Letters database for “Thailand” and the date “02/27/2024”)
- BioAsia Worldwide (Malaysia) — February 2025
- (For the original FDA letter, search the FDA Warning Letters database for “BioAsia Worldwide” and the date “02/2025”)
For the most authoritative and up-to-date versions, always use the FDA Warning Letters database and search by company name and date.
Causal Reasoning: A Transformative Approach to Root Cause Analysis
Energy Safety Canada recently published a white paper on causal reasoning that offers valuable insights for quality professionals across industries. As someone who has spent decades examining how we investigate deviations and perform root cause analysis, I found their framework refreshing and remarkably aligned with the challenges we face in pharmaceutical quality. The paper proposes a fundamental shift in how we approach investigations, moving from what they call “negative reasoning” to “causal reasoning” that could significantly improve our ability to prevent recurring issues and drive meaningful improvement.
The Problem with Traditional Root Cause Analysis
Many of us in quality have experienced the frustration of seeing the same types of deviations recur despite thorough investigations and seemingly robust CAPAs. The Energy Safety Canada white paper offers a compelling explanation for this phenomenon: our investigations often focus on what did not happen rather than what actually occurred.
This approach, which the authors term “negative reasoning,” leads investigators to identify counterfactuals-things that did not occur, such as “operators not following procedures” or “personnel not stopping work when they should have”. The problem is fundamental: what was not happening cannot create the outcomes we experienced. As the authors aptly state, these counterfactuals “exist only in retrospection and never actually influenced events,” yet they dominate many of our investigation conclusions.
This insight resonates strongly with what I’ve observed in pharmaceutical quality. Six years ago the MHRA’s 2019 citation of 210 companies for inadequate root cause analysis and CAPA development – including 6 critical findings – takes on renewed significance in light of Sanofi’s 2025 FDA warning letter. While most cited organizations likely believed their investigation processes were robust (as Sanofi presumably did before their contamination failures surfaced), these parallel cases across regulatory bodies and years expose a persistent industry-wide disconnect between perceived and actual investigation effectiveness. These continued failures exemplify how superficial root cause analysis creates dangerous illusions of control – precisely the systemic flaw the MHRA data highlighted six years prior.
Negative Reasoning vs. Causal Reasoning: A Critical Distinction
The white paper makes a distinction that I find particularly valuable: negative reasoning seeks to explain outcomes based on what was missing from the system, while causal reasoning looks for what was actually present or what happened. This difference may seem subtle, but it fundamentally changes the nature and outcomes of our investigations.
When we use negative reasoning, we create what the white paper calls “an illusion of cause without being causal”. We identify things like “failure to follow procedures” or “inadequate risk assessment,” which may feel satisfying but don’t explain why those conditions existed in the first place. These conclusions often lead to generic corrective actions that fail to address underlying issues.
In contrast, causal reasoning requires statements that have time, place, and magnitude. It focuses on what was necessary and sufficient to create the effect, building a logically tight cause-and-effect diagram. This approach helps reveal how work is actually done rather than comparing reality to an imagined ideal.
This distinction parallels the gap between “work-as-imagined” (the black line) and “work-as-done” (the blue line). Too often, our investigations focus only on deviations from work-as-imagined without trying to understand why work-as-done developed differently.
A Tale of Two Analyses: The Power of Causal Reasoning
The white paper presents a compelling case study involving a propane release and operator injury that illustrates the difference between these two approaches. When initially analyzed through negative reasoning, investigators concluded the operator:
- Used an improper tool
- Deviated from good practice
- Failed to recognize hazards
- Failed to learn from past experiences
These conclusions placed blame squarely on the individual and led leadership to consider terminating the operator.
However, when the same incident was examined through causal reasoning, a different picture emerged:
- The operator used the pipe wrench because it was available at the pump specifically for this purpose
- The pipe wrench had been deliberately left at that location because operators knew the valve was hard to close
- The operator acted quickly because he perceived a risk to the plant and colleagues
- Leadership had actually endorsed this workaround four years earlier during a turnaround
This causally reasoned analysis revealed that what appeared to be an individual failure was actually a system-level issue that had been normalized over time. Rather than punishing the operator, leadership recognized their own role in creating the conditions for the incident and implemented systemic improvements.
This example reminded me of our discussions on barrier analysis, where we examine barriers that failed, weren’t used, or didn’t exist. But causal reasoning takes this further by exploring why those conditions existed in the first place, creating a much richer understanding of how work actually happens.
First 24 Hours: Where Causal Reasoning Meets The Golden Day
In my recent post on “The Golden Start to a Deviation Investigation,” I emphasized how critical the first 24 hours are after discovering a deviation. This initial window represents our best opportunity to capture accurate information and set the stage for a successful investigation. The Energy Safety Canada white paper complements this concept perfectly by providing guidance on how to use those critical hours effectively.
When we apply causal reasoning during these early stages, we focus on collecting specific, factual information about what actually occurred rather than immediately jumping to what should have happened. This means documenting events with specificity (time, place, magnitude) and avoiding premature judgments about deviations from procedures or expectations.
As I’ve previously noted, clear and precise problem definition forms the foundation of any effective investigation. Causal reasoning enhances this process by ensuring we document using specific, factual language that describes what occurred rather than what didn’t happen. This creates a much stronger foundation for the entire investigation.
Beyond Human Error: System Thinking and Leadership’s Role
One of the most persistent challenges in our field is the tendency to attribute events to “human error.” As I’ve discussed before, when human error is suspected or identified as the cause, this should be justified only after ensuring that process, procedural, or system-based errors have not been overlooked. The white paper reinforces this point, noting that human actions and decisions are influenced by the system in which people work.
In fact, the paper presents a hierarchy of causes that resonates strongly with systems thinking principles I’ve advocated for previously. Outcomes arise from physical mechanisms influenced by human actions and decisions, which are in turn governed by systemic factors. If we only address physical mechanisms or human behaviors without changing the system, performance will eventually migrate back to where it has always been.
This connects directly to what I’ve written about quality culture being fundamental to providing quality. The white paper emphasizes that leadership involvement is directly correlated with performance improvement. When leaders engage to set conditions and provide resources, they create an environment where investigations can reveal systemic issues rather than just identify procedural deviations or human errors.
Implementing Causal Reasoning in Pharmaceutical Quality
For pharmaceutical quality professionals looking to implement causal reasoning in their investigation processes, I recommend starting with these practical steps:
1. Develop Investigator Competencies
As I’ve discussed in my analysis of Sanofi’s FDA warning letter, having competent investigators is crucial. Organizations should:
- Define required competencies for investigators
- Provide comprehensive training on causal reasoning techniques
- Implement mentoring programs for new investigators
- Regularly assess and refresh investigator skills
2. Shift from Counterfactuals to Causal Statements
Review your recent investigations and look for counterfactual statements like “operators did not follow the procedure.” Replace these with causal statements that describe what actually happened and why it made sense to the people involved at the time.
3. Implement a Sponsor-Driven Approach
The white paper emphasizes the importance of investigation sponsors (otherwise known as Area Managers) who set clear conditions and expectations. This aligns perfectly with my belief that quality culture requires alignment between top management behavior and quality system philosophy. Sponsors should:
- Clearly define the purpose and intent of investigations
- Specify that a causal reasoning orientation should be used
- Provide resources and access needed to find data and translate it into causes
- Remain engaged throughout the investigation process

4. Use Structured Causal Analysis Tools
While the M-based frameworks I’ve discussed previously (4M, 5M, 6M) remain valuable for organizing contributing factors, they should be complemented with tools that support causal reasoning. The Cause-Consequence Analysis (CCA) I described in a recent post offers one such approach, combining elements of fault tree analysis and event tree analysis to provide a holistic view of risk scenarios.
From Understanding to Improvement
The Energy Safety Canada white paper’s emphasis on causal reasoning represents a valuable contribution to how we think about investigations across industries. For pharmaceutical quality professionals, this approach offers a way to move beyond compliance-focused investigations to truly understand how our systems operate and how to improve them.
As the authors note, “The capacity for an investigation to improve performance is dependent on the type of reasoning used by investigators”. By adopting causal reasoning, we can build investigations that reveal how work actually happens rather than simply identifying deviations from how we imagine it should happen.
This approach aligns perfectly with my long-standing belief that without a strong quality culture, people will not be ready to commit and involve themselves fully in building and supporting a robust quality management system. Causal reasoning creates the transparency and learning that form the foundation of such a culture.
I encourage quality professionals to download and read the full white paper, reflect on their current investigation practices, and consider how causal reasoning might enhance their approach to understanding and preventing deviations. The most important questions to consider are:
- Do your investigation conclusions focus on what didn’t happen rather than what did?
- How often do you identify “human error” without exploring the system conditions that made that error likely?
- Are your leaders engaged as sponsors who set conditions for successful investigations?
- What barriers exist in your organization that prevent learning from events?
As we continue to evolve our understanding of quality and safety, approaches like causal reasoning offer valuable tools for creating the transparency needed to navigate complexity and drive meaningful improvement.
Navigating VUCA and BANI: Building Quality Systems for a Chaotic World
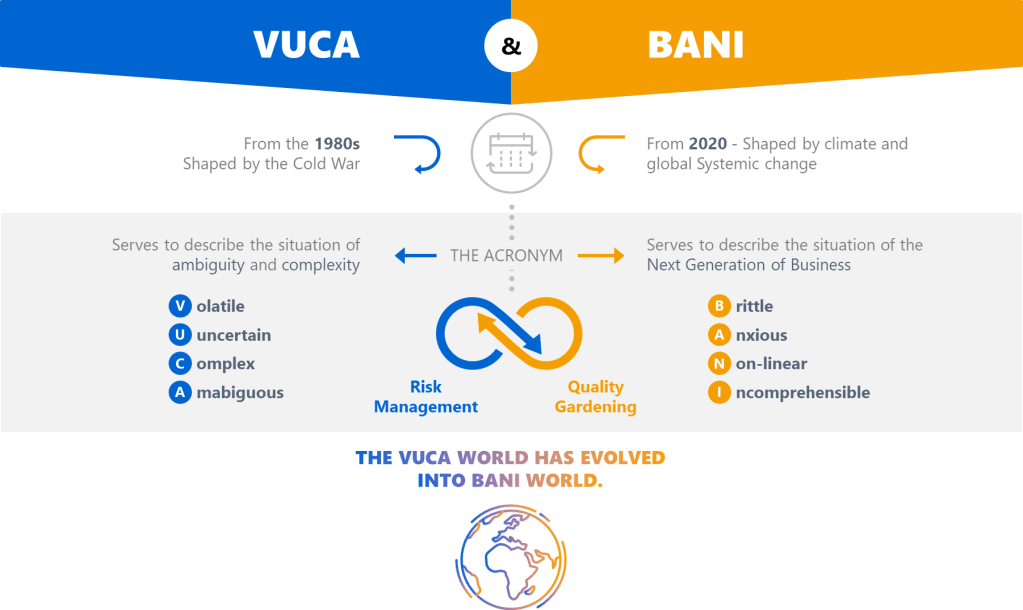
The quality management landscape has always been a battlefield of competing priorities, but today’s environment demands more than just compliance-it requires systems that thrive in chaos. For years, frameworks like VUCA (Volatility, Uncertainty, Complexity, Ambiguity) have dominated discussions about organizational resilience. But as the world fractures into what Jamais Cascio terms a BANI reality (Brittle, Anxious, Non-linear, Incomprehensible), our quality systems must evolve beyond 20th-century industrial thinking. Drawing from my decade of dissecting quality systems on Investigations of a Dog, let’s explore how these frameworks can inform modern quality management systems (QMS) and drive maturity.
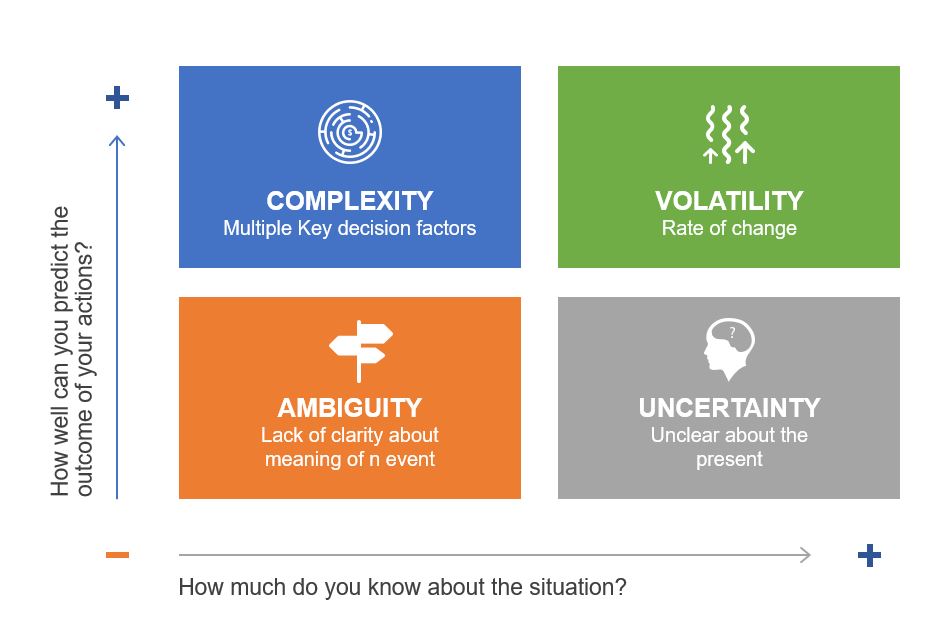
VUCA: A Checklist, Not a Crutch
VUCA entered the lexicon as a military term, but its adoption by businesses has been fraught with misuse. As I’ve argued before, treating VUCA as a single concept is a recipe for poor decisions. Each component demands distinct strategies:
Volatility ≠ Complexity
Volatility-rapid, unpredictable shifts-calls for adaptive processes. Think of commodity markets where prices swing wildly. In pharma, this mirrors supply chain disruptions. The solution isn’t tighter controls but modular systems that allow quick pivots without compromising quality. My post on operational stability highlights how mature systems balance flexibility with consistency.
Ambiguity ≠ Uncertainty
Ambiguity-the “gray zones” where cause-effect relationships blur-is where traditional QMS often stumble. As I noted in Dealing with Emotional Ambivalence, ambiguity aversion leads to over-standardization. Instead, build experimentation loops into your QMS. For example, use small-scale trials to test contamination controls before full implementation.
BANI: The New Reality Check
Cascio’s BANI framework isn’t just an update to VUCA-it’s a wake-up call. Let’s break it down through a QMS lens:
Brittle Systems Break Without Warning
The FDA’s Quality Management Maturity (QMM) program emphasizes that mature systems withstand shocks. But brittleness lurks in overly optimized processes. Consider a validation program that relies on a single supplier: efficient, yes, but one disruption collapses the entire workflow. My maturity model analysis shows that redundancy and diversification are non-negotiable in brittle environments.
Anxiety Demands Psychological Safety
Anxiety isn’t just an individual burden, it’s systemic. In regulated industries, fear of audits often drives document hoarding rather than genuine improvement. The key lies in cultural excellence, where psychological safety allows teams to report near-misses without blame.
Non-Linear Cause-Effect Upends Root Cause Analysis
Traditional CAPA assumes linearity: find the root cause, apply a fix. But in a non-linear world, minor deviations cascade unpredictably. We need to think more holistically about problem solving.
Incomprehensibility Requires Humility
When even experts can’t grasp full system interactions, transparency becomes strategic. Adopt open-book quality metrics to share real-time data across departments. Cross-functional reviews expose blind spots.
Building a BANI-Ready QMS
From Documents to Living Systems
Traditional QMS drown in documents that “gather dust” (Documents and the Heart of the Quality System). Instead, model your QMS as a self-adapting organism:
- Use digital twins to simulate disruptions
- Embed risk-based decision trees in SOPs
- Replace annual reviews with continuous maturity assessments
Maturity Models as Navigation Tools
A maturity model framework maps five stages from reactive to anticipatory. Utilizing a Maturity model for quality planning help prepare for what might happen.
Operational Stability as the Keystone
The House of Quality model positions operational stability as the bridge between culture and excellence. In BANI’s brittle world, stability isn’t rigidity-it’s dynamic equilibrium. For example, a plant might maintain ±1% humidity control not by tightening specs but by diversifying HVAC suppliers and using real-time IoT alerts.
The Path Forward
VUCA taught us to expect chaos; BANI forces us to surrender the illusion of control. For quality leaders, this means:
- Resist checklist thinking: VUCA’s four elements aren’t boxes to tick but lenses to sharpen focus.
- Embrace productive anxiety: As I wrote in Ambiguity, discomfort drives innovation when channeled into structured experimentation.
- Invest in sensemaking: Tools like Quality Function Deployment help teams contextualize fragmented data.
The future belongs to quality systems that don’t just survive chaos but harness it. As Cascio reminds us, the goal isn’t to predict the storm but to learn to dance in the rain.
For deeper dives into these concepts, explore my series on VUCA and Quality Systems.
