In today’s pharmaceutical and biopharmaceutical manufacturing landscape, operational agility through multi-purpose equipment utilization has evolved from competitive advantage to absolute necessity. The industry’s shift toward personalized medicines, advanced therapies, and accelerated development timelines demands manufacturing systems capable of rapid, validated transitions between different processes and products. However, this operational flexibility introduces complex regulatory challenges that extend well beyond basic compliance considerations.
As pharmaceutical professionals navigate this dynamic environment, equipment qualification emerges as the cornerstone of a robust quality system—particularly when implementing multi-purpose manufacturing strategies with single-use technologies. Having guided a few organizations through these qualification challenges over the past decade, I’ve observed a fundamental misalignment between regulatory expectations and implementation practices that creates unnecessary compliance risk.
In this post, I want to explore strategies for qualifying equipment across different processes, with particular emphasis on leveraging single-use technologies to simplify transitions while maintaining robust compliance. We’ll explore not only the regulatory framework but the scientific rationale behind qualification requirements when operational parameters change. By implementing these systematized approaches, organizations can simultaneously satisfy regulatory expectations and enhance operational efficiency—transforming compliance activities from burden to strategic advantage.
The Fundamentals: Equipment Requalification When Parameters Change
When introducing a new process or expanding operational parameters, a fundamental GMP requirement applies: equipment qualification ranges must undergo thorough review and assessment. Regulatory guidance is unambiguous on this point: Whenever a new process is introduced the qualification ranges should be reviewed. If equipment has been qualified over a certain range and is required to operate over a wider range than before, prior to use it should be re-qualified over the wider range.
This requirement stems from the scientific understanding that equipment performance characteristics can vary significantly across different operational ranges. Temperature control systems that maintain precise stability at 37°C may exhibit unacceptable variability at 4°C. Mixing systems designed for aqueous formulations may create detrimental shear forces when processing more viscous products. Control algorithms optimized for specific operational setpoints might perform unpredictably at the extremes of their range.
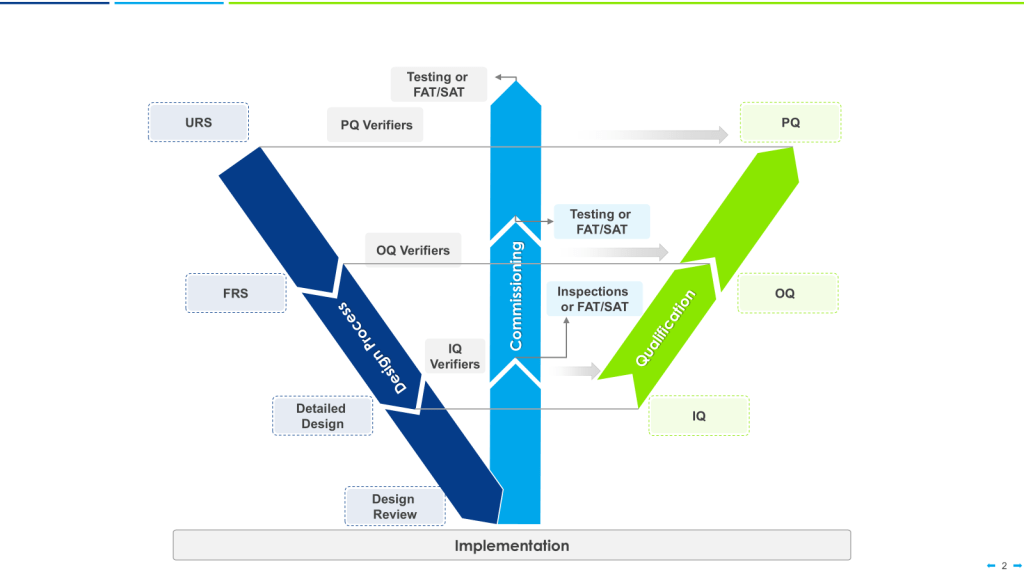
There are a few risk-based models of verification, such as the 4Q qualification model—consisting of Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ)— or the W-Model which can provide a structured framework for evaluating equipment performance across varied operating conditions. These widely accepted approaches ensures comprehensive verification that equipment will consistently produce products meeting quality requirements. For multi-purpose equipment specifically, the Performance Qualification phase takes on heightened importance as it confirms consistent performance under varied processing conditions.
I cannot stress the importance of risk based approach of ASTM E2500 here which emphasizes a flexible verification strategy focused on critical aspects that directly impact product quality and patient safety. ASTM E2500 integrates several key principles that transform equipment qualification from a documentation exercise to a scientific endeavor:
Risk-based approach: Verification activities focus on critical aspects with the potential to affect product quality, with the level of effort and documentation proportional to risk. As stated in the standard, “The evaluation of risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient”.
- Science-based decisions: Product and process information, including critical quality attributes (CQAs) and critical process parameters (CPPs), drive verification strategies. This ensures that equipment verification directly connects to product quality requirements.
- Quality by Design integration: Critical aspects are designed into systems during development rather than tested in afterward, shifting focus from testing quality to building it in from the beginning.
- Subject Matter Expert (SME) leadership: Technical experts take leading roles in verification activities appropriate to their areas of expertise.
- Good Engineering Practice (GEP) foundation: Engineering principles and practices underpin all specification, design, and verification activities, creating a more technically robust approach to qualification
Organizations frequently underestimate the technical complexity and regulatory significance of equipment requalification when operational parameters change. The common misconception that equipment qualified for one process can simply be repurposed for another without formal assessment creates not only regulatory vulnerability but tangible product quality risks. Each expansion of operational parameters requires systematic evaluation of equipment capabilities against new requirements—a scientific approach rather than merely a documentation exercise.
Single-Use Systems: Revolutionizing Multi-Purpose Manufacturing
Single-use technologies (SUT) have fundamentally transformed how organizations approach process transitions in biopharmaceutical manufacturing. By eliminating cleaning validation requirements and dramatically reducing cross-contamination risks, these systems enable significantly more rapid equipment changeovers between different products and processes. However, this operational advantage comes with distinct qualification considerations that require specialized expertise.
The qualification approach for single-use systems differs fundamentally from traditional stainless equipment due to the redistribution of quality responsibility across the supply chain. I conceptualize SUT validation as operating across three interconnected domains, each requiring distinct validation strategies:
- Process operation validation: This domain focuses on the actual processing parameters, aseptic operations, product hold times, and process closure requirements specific to each application. For multi-purpose equipment, this validation must address each process’s unique requirements while ensuring compatibility across all intended applications.
- Component manufacturing validation: This domain centers on the supplier’s quality systems for producing single-use components, including materials qualification, manufacturing controls, and sterilization validation. For organizations implementing multi-purpose strategies, supplier validation becomes particularly critical as component properties must accommodate all intended processes.
- Supply chain process validation: This domain ensures consistent quality and availability of single-use components throughout their lifecycle. For multi-purpose applications, supply chain robustness takes on heightened importance as component variability could affect process consistency across different applications.
This redistribution of quality responsibility creates both opportunities and challenges. Organizations can leverage comprehensive vendor validation packages to accelerate implementation, reducing qualification burden compared to traditional equipment. However, this necessitates implementing unusually robust supplier qualification programs that thoroughly evaluate manufacturer quality systems, change control procedures, and extractables/leachables studies applicable across all intended process conditions.
When qualifying single-use systems for multi-purpose applications, material science considerations become paramount. Each product formulation may interact differently with single-use materials, potentially affecting critical quality attributes through mechanisms like protein adsorption, leachable compound introduction, or particulate generation. These product-specific interactions must be systematically evaluated for each application, requiring specialized analytical capabilities and scientifically sound acceptance criteria.
Proving Effective Process Transitions Without Compromising Quality
For equipment designed to support multiple processes, qualification must definitively demonstrate the system can transition effectively between different applications without compromising performance or product quality. This demonstration represents a frequent focus area during regulatory inspections, where the integrity of product changeovers is routinely scrutinized.
When utilizing single-use systems, the traditional cleaning validation burden is substantially reduced since product-contact components are replaced between processes. However, several critical elements still require rigorous qualification:
Changeover procedures must be meticulously documented with detailed instructions for disassembly, disposal of single-use components, assembly of new components, and verification steps. These procedures should incorporate formal engineering assessments of mechanical interfaces to prevent connection errors during reassembly. Verification protocols should include explicit acceptance criteria for visual inspection of non-disposable components and connection points, with particular attention to potential entrapment areas where residual materials might accumulate.
Product-specific impact assessments represent another critical element, evaluating potential interactions between product formulations and equipment materials. For single-use systems specifically, these assessments should include:
- Adsorption potential based on product molecular properties, including molecular weight, charge distribution, and hydrophobicity
- Extractables and leachables unique to each formulation, with particular attention to how process conditions (temperature, pH, solvent composition) might affect extraction rates
- Material compatibility across the full range of process conditions, including extreme parameter combinations that might accelerate degradation
- Hold time limitations considering both product quality attributes and single-use material integrity under process-specific conditions
Process parameter verification provides objective evidence that critical parameters remain within acceptable ranges during transitions. This verification should include challenging the system at operational extremes with each product formulation, not just at nominal settings. For temperature-controlled processes, this might include verification of temperature recovery rates after door openings or evaluation of temperature distribution patterns under different loading configurations.
An approach I’ve found particularly effective is conducting “bracketing studies” that deliberately test worst-case combinations of process parameters with different product formulations. These studies specifically evaluate boundary conditions where performance limitations are most likely to manifest, such as minimum/maximum temperatures combined with minimum/maximum agitation rates. This provides scientific evidence that the equipment can reliably handle transitions between the most challenging operating conditions without compromising performance.
When applying the W-model approach to validation, special attention should be given to the verification stages for multi-purpose equipment. Each verification step must confirm not only that the system meets individual requirements but that it can transition seamlessly between different requirement sets without compromising performance or product quality.
Developing Comprehensive User Requirement Specifications
The foundation of effective equipment qualification begins with meticulously defined User Requirement Specifications (URS). For multi-purpose equipment, URS development requires exceptional rigor as it must capture the full spectrum of intended uses while establishing clear connections to product quality requirements.
A URS for multi-purpose equipment should include:
Comprehensive operational ranges for all process parameters across all intended applications. Rather than simply listing individual setpoints, the URS should define the complete operating envelope required for all products, including normal operating ranges, alert limits, and action limits. For temperature-controlled processes, this should specify not only absolute temperature ranges but stability requirements, recovery time expectations, and distribution uniformity standards across varied loading scenarios.
Material compatibility requirements for all product formulations, particularly critical for single-use technologies where material selection significantly impacts extractables profiles. These requirements should reference specific material properties (rather than just general compatibility statements) and establish explicit acceptance criteria for compatibility studies. For pH-sensitive processes, the URS should define the acceptable pH range for all contact materials and specify testing requirements to verify material performance across that range.
Changeover requirements detailing maximum allowable transition times, verification methodologies, and product-specific considerations. This should include clearly defined acceptance criteria for changeover verification, such as visual inspection standards, integrity testing parameters for assembled systems, and any product-specific testing requirements to ensure residual clearance.
Future flexibility considerations that build in reasonable operational margins beyond current requirements to accommodate potential process modifications without complete requalification. This forward-looking approach avoids the common pitfall of qualifying equipment for the minimum necessary range, only to require requalification when minor process adjustments are implemented.
Explicit connections between equipment capabilities and product Critical Quality Attributes (CQAs), demonstrating how equipment performance directly impacts product quality for each application. This linkage establishes the scientific rationale for qualification requirements, helping prioritize testing efforts around parameters with direct impact on product quality.
The URS should establish unambiguous, measurable acceptance criteria that will be used during qualification to verify equipment performance. These criteria should be specific, testable, and directly linked to product quality requirements. For temperature-controlled processes, rather than simply stating “maintain temperature of X°C,” specify “maintain temperature of X°C ±Y°C as measured at multiple defined locations under maximum and minimum loading conditions, with recovery to setpoint within Z minutes after a door opening event.”
Qualification Testing Methodologies: Beyond Standard Approaches
Qualifying multi-purpose equipment requires more sophisticated testing strategies than traditional single-purpose equipment. The qualification protocols must verify performance not only at standard operating conditions but across the full operational spectrum required for all intended applications.
Installation Qualification (IQ) Considerations
For multi-purpose equipment using single-use systems, IQ should verify proper integration of disposable components with permanent equipment, including:
- Comprehensive documentation of material certificates for all product-contact components, with particular attention to material compatibility with all intended process conditions
- Verification of proper connections between single-use assemblies and fixed equipment, including mechanical integrity testing of connection points under worst-case pressure conditions
- Confirmation that utilities meet specifications across all intended operational ranges, not just at nominal settings
- Documentation of system configurations for each process the equipment will support, including component placement, connection arrangements, and control system settings
- Verification of sensor calibration across the full operational range, with particular attention to accuracy at the extremes of the required range
The IQ phase should be expanded for multi-purpose equipment to include verification that all components and instrumentation are properly installed to support each intended process configuration. When additional processes are added after the fact a retrospective fit-for-purpose assessment should be conducted and gaps addressed.
Operational Qualification (OQ) Approaches
OQ must systematically challenge the equipment across the full range of operational parameters required for all processes:
- Testing at operational extremes, not just nominal setpoints, with particular attention to parameter combinations that represent worst-case scenarios
- Challenge testing under boundary conditions for each process, including maximum/minimum loads, highest/lowest processing rates, and extreme parameter combinations
- Verification of control system functionality across all operational ranges, including all alarms, interlocks, and safety features specific to each process
- Assessment of performance during transitions between different parameter sets, evaluating control system response during significant setpoint changes
- Robustness testing that deliberately introduces disturbances to evaluate system recovery capabilities under various operating conditions
For temperature-controlled equipment specifically, OQ should verify temperature accuracy and stability not only at standard operating temperatures but also at the extremes of the required range for each process. This should include assessment of temperature distribution patterns under different loading scenarios and recovery performance after system disturbances.
Performance Qualification (PQ) Strategies
PQ represents the ultimate verification that equipment performs consistently under actual production conditions:
- Process-specific PQ protocols demonstrating reliable performance with each product formulation, challenging the system with actual production-scale operations
- Process simulation tests using actual products or qualified substitutes to verify that critical quality attributes are consistently achieved
- Multiple assembly/disassembly cycles when using single-use systems to demonstrate reliability during process transitions
- Statistical evaluation of performance consistency across multiple runs, establishing confidence intervals for critical process parameters
- Worst-case challenge tests that combine boundary conditions for multiple parameters simultaneously
For organizations implementing the W-model, the enhanced verification loops in this approach provide particular value for multi-purpose equipment, establishing robust evidence of equipment performance across varied operating conditions and process configurations.
Fit-for-Purpose Assessment Table: A Practical Tool
When introducing a new platform product to existing equipment, a systematic assessment is essential. The following table provides a comprehensive framework for evaluating equipment suitability across all relevant process parameters.
| Column | Instructions for Completion |
|---|---|
| Critical Process Parameter (CPP) | List each process parameter critical to product quality or process performance. Include all parameters relevant to the unit operation (temperature, pressure, flow rate, mixing speed, pH, conductivity, etc.). Each parameter should be listed on a separate row. Parameters should be specific and measurable, not general capabilities. |
| Current Qualified Range | Document the validated operational range from the existing equipment qualification documents. Include both the absolute range limits and any validated setpoints. Specify units of measurement. Note if the parameter has alerting or action limits within the qualified range. Reference the specific qualification document and section where this range is defined. |
| New Required Range | Specify the range required for the new platform product based on process development data. Include target setpoint and acceptable operating range. Document the source of these requirements (e.g., process characterization studies, technology transfer documents, risk assessments). Specify units of measurement identical to those used in the Current Qualified Range column for direct comparison. |
| Gap Analysis | Quantitatively assess whether the new required range falls completely within the current qualified range, partially overlaps, or falls completely outside. Calculate and document the specific gap (numerical difference) between ranges. If the new range extends beyond the current qualified range, specify in which direction (higher/lower) and by how much. If completely contained within the current range, state “No Gap Identified.” |
| Equipment Capability Assessment | Evaluate whether the equipment has the physical/mechanical capability to operate within the new required range, regardless of qualification status. Review equipment specifications from vendor documentation to confirm design capabilities. Consult with equipment vendors if necessary to confirm operational capabilities not explicitly stated in documentation. Document any physical limitations that would prevent operation within the required range. |
| Risk Assessment | Perform a risk assessment evaluating the potential impact on product quality, process performance, and equipment integrity when operating at the new parameters. Use a risk ranking approach (High/Medium/Low) with clear justification. Consider factors such as proximity to equipment design limits, impact on material compatibility, effect on equipment lifespan, and potential failure modes. Reference any formal risk assessment documents that provide more detailed analysis. |
| Automation Capability | Assess whether the current automation system can support the new required parameter ranges. Evaluate control algorithm suitability, sensor ranges and accuracy across the new parameters, control loop performance at extreme conditions, and data handling capacity. Identify any required software modifications, control strategy updates, or hardware changes to support the new operating ranges. Document testing needed to verify automation performance across the expanded ranges. |
| Alarm Strategy | Define appropriate alarm strategies for the new parameter ranges, including warning and critical alarm setpoints. Establish allowable excursion durations before alarm activation for dynamic parameters. Compare new alarm requirements against existing configured alarms, identifying gaps. Evaluate alarm prioritization and ensure appropriate operator response procedures exist for new or modified alarms. Consider nuisance alarm potential at expanded operating ranges and develop mitigation strategies. |
| Required Modifications | Document any equipment modifications, control system changes, or additional components needed to achieve the new required range. Include both hardware and software modifications. Estimate level of effort and downtime required for implementation. If no modifications are needed, explicitly state “No modifications required.” |
| Testing Approach | Outline the specific qualification approach for verifying equipment performance within the new required range. Define whether full requalification is needed or targeted testing of specific parameters is sufficient. Specify test methodologies, sampling plans, and duration of testing. Detail how worst-case conditions will be challenged during testing. Reference any existing protocols that will be leveraged or modified. For single-use systems, address how single-use component integration will be verified. |
| Acceptance Criteria | Define specific, measurable acceptance criteria that must be met to demonstrate equipment suitability. Criteria should include parameter accuracy, stability, reproducibility, and control precision. Specify statistical requirements (e.g., capability indices) if applicable. Ensure criteria address both steady-state operation and response to disturbances. For multi-product equipment, include criteria related to changeover effectiveness. |
| Documented Evidence Required | List specific documentation required to support the fit-for-purpose determination. Include qualification protocols/reports, engineering assessments, vendor statements, material compatibility studies, and historical performance data. For single-use components, specify required vendor documentation (e.g., extractables/leachables studies, material certificates). Identify whether existing documentation is sufficient or new documentation is needed. |
| Impact on Concurrent Products | Assess how qualification activities or equipment modifications for the new platform product might impact other products currently manufactured using the same equipment. Evaluate schedule conflicts, equipment availability, and potential changes to existing qualified parameters. Document strategies to mitigate any negative impacts on existing production. |
Implementation Guidelines
The Equipment Fit-for-Purpose Assessment Table should be completed through structured collaboration among cross-functional stakeholders, with each Critical Process Parameter (CPP) evaluated independently while considering potential interaction effects.
- Form a cross-functional team including process engineering, validation, quality assurance, automation, and manufacturing representatives. For technically complex assessments, consider including representatives from materials science and analytical development to address product-specific compatibility questions.
- Start with comprehensive process development data to clearly define the required operational ranges for the new platform product. This should include data from characterization studies that establish the relationship between process parameters and Critical Quality Attributes, enabling science-based decisions about qualification requirements.
- Review existing qualification documentation to determine current qualified ranges and identify potential gaps. This review should extend beyond formal qualification reports to include engineering studies, historical performance data, and vendor technical specifications that might provide additional insights about equipment capabilities.
- Evaluate equipment design capabilities through detailed engineering assessment. This should include review of design specifications, consultation with equipment vendors, and potentially non-GMP engineering runs to verify equipment performance at extended parameter ranges before committing to formal qualification activities.
- Conduct parameter-specific risk assessments for identified gaps, focusing on potential impact to product quality. These assessments should apply structured methodologies like FMEA (Failure Mode and Effects Analysis) to quantify risks and prioritize qualification efforts based on scientific rationale rather than arbitrary standards.
- Develop targeted qualification strategies based on gap analysis and risk assessment results. These strategies should pay particular attention to Performance Qualification under process-specific conditions.
- Generate comprehensive documentation to support the fit-for-purpose determination, creating an evidence package that would satisfy regulatory scrutiny during inspections. This documentation should establish clear scientific rationale for all decisions, particularly when qualification efforts are targeted rather than comprehensive.
The assessment table should be treated as a living document, updated as new information becomes available throughout the implementation process. For platform products with established process knowledge, leveraging prior qualification data can significantly streamline the assessment process, focusing resources on truly critical parameters rather than implementing blanket requalification approaches.
When multiple parameters show qualification gaps, a science-based prioritization approach should guide implementation strategy. Parameters with direct impact on Critical Quality Attributes should receive highest priority, followed by those affecting process consistency and equipment integrity. This prioritization ensures that qualification efforts address the most significant risks first, creating the greatest quality benefit with available resources.
Building a Robust Multi-Purpose Equipment Strategy
As biopharmaceutical manufacturing continues evolving toward flexible, multi-product facilities, qualification of multi-purpose equipment represents both a regulatory requirement and strategic opportunity. Organizations that develop expertise in this area position themselves advantageously in an increasingly complex manufacturing landscape, capable of rapidly introducing new products while maintaining unwavering quality standards.
The systematic assessment approaches outlined in this article provide a scientific framework for equipment qualification that satisfies regulatory expectations while optimizing operational efficiency. By implementing tools like the Fit-for-Purpose Assessment Table and leveraging a risk-based validation model, organizations can navigate the complexities of multi-purpose equipment qualification with confidence.
Single-use technologies offer particular advantages in this context, though they require specialized qualification considerations focusing on supplier quality systems, material compatibility across different product formulations, and supply chain robustness. Organizations that develop systematic approaches to these considerations can fully realize the benefits of single-use systems while maintaining robust compliance.
The most successful organizations in this space recognize that multi-purpose equipment qualification is not merely a regulatory obligation but a strategic capability that enables manufacturing agility. By building expertise in this area, biopharmaceutical manufacturers position themselves to rapidly introduce new products while maintaining the highest quality standards—creating a sustainable competitive advantage in an increasingly dynamic market.