Classification of change controls within change management is a common and widely accepted best practice. It stems from the requirement that change proposals as assessed from a risk perspective, where:
- the level of rigor, effort and documentation is commensurate with the level of risk,
- the risk assessments adequately evaluate the potential risks and benefits of changes to product quality, safety and efficacy, and
- those risk assessments consider the potential risks and benefits to other products, processes and systems.
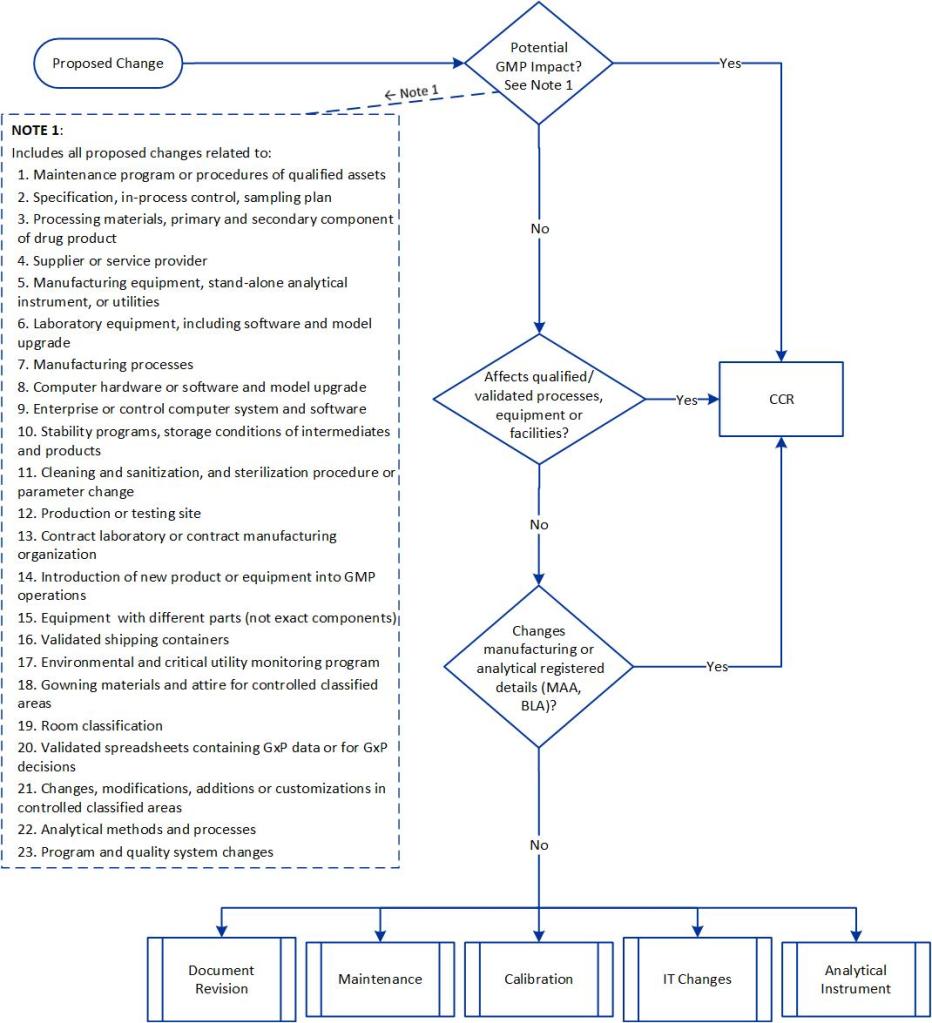
Classification for GMP/GDP changes itself is not a requirement, it is a guidance, best found in the PIC/S Recommendation “How to Evaluate and Demonstrate the Effectiveness of a Pharmaceutical Quality System in relation to Risk-based Change Management” (PI 054-1) which states in section 5.2 “Change Management procedures often require a risk-based classification (e.g. critical, major, minor) to be assigned to proposed changes as well as an impact assessment to be performed. The latter routinely determines the potential impacts of the proposed change on various items, such as product quality, documentation, cleaning, maintenance, regulatory compliance, etc. In some cases, especially for simple and minor/low risk changes, an impact assessment is sufficient to document the risk-based rationale for a change without the use of more formal risk assessment tools or approaches.”
The PIC/S tells us that these categories drive the amount of rigor a change control requires, which is a great reason to have them. We spend time creating and confirming our categories, and then we only need to perform more rigorous risk assessments on the big changes.
How should we build this risk-based classification system? There are four criteria that drive this:
- Potential regulatory impact
- Potential impact on the qualified and validated state
- Potential impact on the ability to disposition and ship product
- Complexity
I tend to use only two categories, defined like this:
Major has Significant Impact: Changes that have a considerable potential impact on the process, product quality, safety, or regulatory status.
Minor has Limited Impact: Changes that have minimal or no significant impact on the process, product quality, safety, or regulatory status.

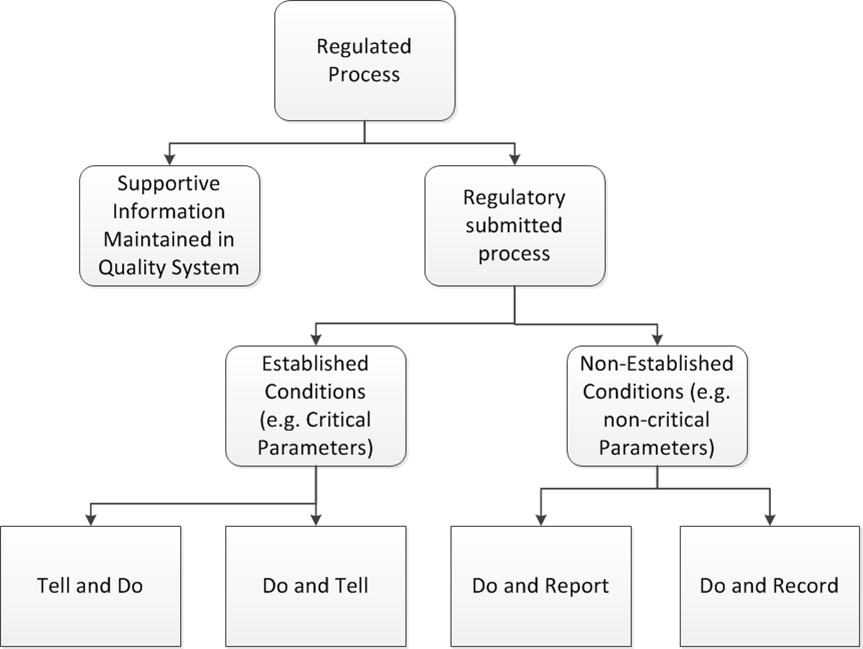
For regulatory impact, it really is as easy as dividing things into the four categories. “Do, Report, and Do and Record are minors. “Do and Tell” are majors, and “Tell and Do are either majors or critical based on how you slice it.
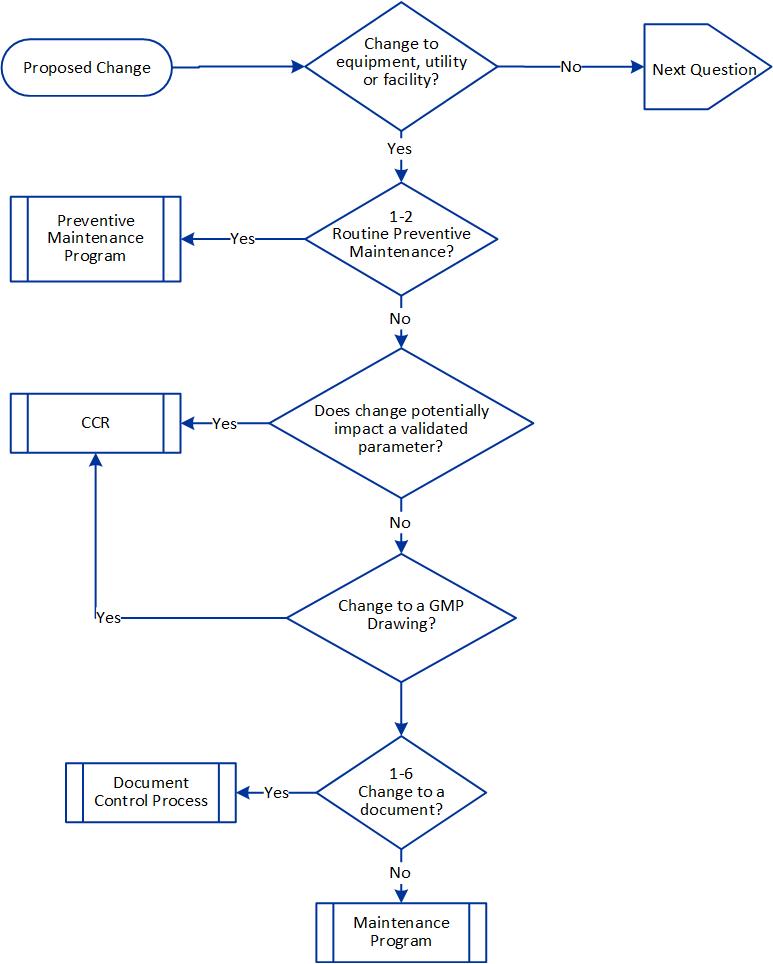
When considering potential validation impact you’ll leverage your process risk assessments and your validated state to determine what is in that bucket. This is why I like a document like an operational control strategy because this tells me exactly what impacts my validated state and I can just it to form this category.
The potential impact on the ability to disposition and ship the product has me looking at what can impact the ability to release and get the product out the door, which is an important aspect of what we do. Remember, a shortage of products is a quality issue.
Complexity looks at how many processes and systems are impacted and how many functions and areas are involved. The more complex, the more formal risk assessment is required. For example, you might use groupings like this:
Low level of complexity
- Requires actions from the change owner and the system owner’s department(s) only
- Impacts 1 system
- <10 document revisions (approximate)
- <2 potential training audiences (approximate)
Higher complexity:
- Requires actions from more than change owner and system owner
- Impacts more than one system
- >10 document revisions
- >2 potential training audiences (approximate)
The where of making the classification also makes a difference. I recommend up front, agreed to by the change owner and quality and it then drives everything. Doing it just before approval really just decides who gets to approve the change control and whether it goes to CCRB or not.
These classifications can be loose guidelines; for example, a table that looks at the first three categories and then by complexity. Your rating depends on whichever Impact or Complexity is higher.
| Impact of Change (regulatory, validation, product) | Complexity of Change | ||
| Minor | No risk to patient as assessed by SISPQ, product, or validated equipment or process AND No regulatory impact | Limited impact to only one system/functional area AND Has defined process for implementation of change. (e.g. all action items are per defined procedures) | |
| Major | Potential impact to patient or product SISPQ or validated equipment or process or compliance | Impacts multiple systems / functional areas OR Has defined process for implementation of change | |
| Critical | High likelihood of impact to patient, product SISPQ or validated equipment or process or compliance | Impacts multiple systems / functional areas OR Implementation activities are not pre-defined or governed by formal internal system | |
Or we could try for something much more specific. The advantage of specific is any change owner can start making the determination. Something like this:
| Change Category | Change Description |
| Manufacturing Processes | In-process labeling |
| Changes to Process Control and Operating Parameters (tightening/shifting) within current batch record (does not impact established conditions) | |
| The addition of in-process or final product samples | |
| Changes to sample volume for in-process or finished product samples | |
| Addition of new ancillary equipment (e.g. no product contact, does not control process steps) to the process | |
| Analytical Methods | Changes to the qualification of a critical reagent (i.e., in-house produced assay standards and controls) |
| Use of an additional new instrument of the identical model and vendor | |
| Change in compendial method to comply with formal updates to compendia, provided it does not involve the widening of system suitability or acceptance criteria | |
| Equipment/instruments calibration, maintenance, and cleaning | |
| Changes to software or validated analytical spreadsheets that do not impact the current validated state of the method | |
| Movement of instruments from one location to another in the same room/lab | |
| Initial validation of analytical spreadsheets for use in calculation of data and results defined by a specific analytical method, provided it does not replace a worksheet in an SOP (if so, this change may be reportable) | |
| Changes to non-critical equipment or materials that allow “or equivalent” in current method, provided method re-validation is not required | |
| Drug Substance or Drug Product Specifications/ Limits | Changes to the sampling plan involving changes to the number of extra samples or amount of sample provided to QC or CMO as appropriate. |
| Changes to the storage and/or shipping conditions of samples (except for stability vials) | |
| Raw Materials/Com ponents | Compendial Specification Changes to meet Compendial updates |
| Non-product contact filters | |
| Vendor increase or decrease in the number of items per shipping container, or the size of the shipping or outer container | |
| Changes to the vendor Certificate of Analysis (format change only) | |
| Changes in recommended expiration date and/or storage conditions of raw material | |
| Finished Goods | Catalog Number changes to components |
| Creation of label at contract manufacturing site for existing presentation (assuming ‘No’ other change to already approved label) | |
| Changing position of pharmacode on leaflet | |
| Computer | When there is no validation impact |
| Facility, Utilities, Systems and Equipment (including Automation) | Equipment/instrument maintenance |
| Decommissioning of equipment not classified as critical equipment | |
| Computer programming that affects non-production equipment | |
| Alarms (i.e., notification system for out of tolerances) | |
| Cleaning and Sanitization of Manufacturing facilities and non-product Contact equipment | |
| Upgrade of Application Software or operating system | |
| Alarm set point changes | |
| Creating user groups and modifying user group privileges | |
| Tuning parameter, adjustment to the gain, reset and rate of a PID controller | |
| Phase or sequence change that does not affect the function and performance | |
| Modifying a phase prompt or message (technical change) | |
| Addition of a graphic, adding or changing a non-static device to a graphic (technical change) | |
| Addition or changing to an interlock/permissive trigger | |
| Changes to alarm paging/notification functionality |
Spend the time on your classification structure. You will use it to:
- Determine level of risk assessment (major yes, minor no)
- Determine approvals (minors can be as simple as change owner and quality)
- Does this change require a CCRB? Only send majors.