Guidances are an interesting part of our job. As a best practice, they show one way to get to the desired end result, but there can be other ways but the presence of guidance can obscure those possibilities. Often times if an agency goes to the level of detail to show you what good looks like you’d be foolish not to try to meet them there. Other times guidance can be a real head scratcher.
Good article, and of interest to the non-lawyers like myself who have to live within the boundaries.
There has been a lot of press lately for the Abbott Nutrition recall of infant formula. Fundamentally this is a colossal failure of our regulatory program, another failure in a long string of failures, and confirmation that the time is now for radical changes in the agency.
The optimist in me hopes that this calamity will drive needed change, as has been the unfortunate history of regulatory change in this country. I’m just not sure I hold enough confidence in Congress to get the job done.
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 3: Premises and Equipment, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 5: Production, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Part II: Basic Requirements for Active Substances used as Starting Materials, (2014)
European Union, Guidelines of 19 March 2015 on the formalized risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use, Official Journal of the European Union, (2015/C 95/02), (2015)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use, (2018)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 3 Manufacture of Radiopharmaceuticals, (2008)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 14 Manufacture of Medicinal Products Derived from Human Blood or Plasma, (2011)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products, (2017)
European Union, Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, Official Journal of the European Union, (2013/C 343/01), (2013),
European Union, Guidelines of 19 March 2015 on principles of Good Distribution Practice of active substances for medicinal products for human use, Official Journal of the European Union, (2015/C 95/01), (2015)
EMA Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (20 November 2014)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, subpart C = Building and Facilities, sec. 211.42 Design and construction features (b), (c)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart F – Production and Process Controls, sec. 211.113 Control of microbial contamination (a), (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart B – Organization and Personnel, sec.211.28 Personnel responsibilities (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control ofComponents and Drug Product Containers and Closures, sec. 211.80 General requirements. (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control of Components and Drug Product Containers and Closures, sec. 211.84 Testing and approval or rejection of components, drug product containers, and closures (d)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart D – Equipment, sec.211.67 Equipment cleaning and maintenance (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart C – Buildings and Facilities, sec. 211.56 Sanitation (c)
U.S. Food & Drug Administration, Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, (2004)
U.S. Food & Drug Administration, Guidance for Industry – Good Manufacturing Practice Considerations for Responding to COVID-19 Infection in Employees in Drug and Biological Products Manufacturing, (2020)
U.S. Food & Drug Administration, Guidance for Industry – Guidance for Industry Non-Penicillin Beta-Lactam Drugs: A CGMP Framework for Preventing Cross Contamination, (2013)
U.S. Food & Drug Administrationn, Guidance for Industry Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act, Draft Guidance. https://www.fda.gov/media/88905/download (accessed Mar 6, 2022)
Pharmaceutical Inspection Co-operation Scheme gmp guide, 2nd targeted consultation document on revision of annex 1
Pharmaceutical Inspection Co-operation Schemepharmaceutical inspection co-operation scheme gmp guide, ps inf 25 2019 (rev. 1) draft, manufacture of advanced therapy medicinal products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, ps inf 26 2019 (rev. 1) draft, manufacture of biological medicinal substances and products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part i), guide to good manufacturing practice for medicinal products part i
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part ii), guide to good manufacturing practice for medicinal products part ii
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (annexes), guide to good manufacturing practice for medicinal products annexes
World Health Organisation, good manufacturing practices for pharmaceutical products: main principles, annex 2, who technical report series 986, 2014,
World Health Organisation, who good manufacturing practices for active pharmaceutical ingredients (bulk drug substances), annex 2, who technical report series 957, 2010
World Health Organisation, points to consider for manufacturers and inspectors: environmental aspects of manufacturing for the prevention of antimicrobial resistance annex 6, who technical report series 1025, 2020
World Health Organisation, WHO good manufacturing practices for sterile pharmaceutical products, annex 6, who technical report series 961, 2011
World Health Organisation, WHO good manufacturing practices for biological products, annex 3, who technical report series 996, 2016
World Health Organisation, WHO good manufacturing practices for the manufacture of investigational pharmaceutical products for clinical trials in humans, annex 7, who technical report series 863, 1996
World Health Organisation, WHO good manufacturing practices for radiopharmaceutical products annex 2, who technical report series 1025, 2020
World Health Organisation, WHO GMP for Pharmaceutical Products containing Hazardous Substances, TRS 957, Annex-3 (2010)
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, Quality Risk Management, Q8 (R2), Pharmaceutical Development, August 2009. https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf (Accessed Mar 06, 2022)
Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
Regulation/Guidance
States
ICH E-6 (R2) Section 4.5.1-4.5.4
4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion.
ICH E3, section 9.6
The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report
21CFR 312.53(vi) (a)
investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.”
21CFR 56.108(a)
IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects.
21 CFR 56.108(b)
“IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.”
45 CFR 46.103(b)(5)
Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB.
FDA Form-1572 (Section 9)
lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.”
A few key regulations and guidances (not meant to be a comprehensive list)
How Protocol Deviations are Implemented
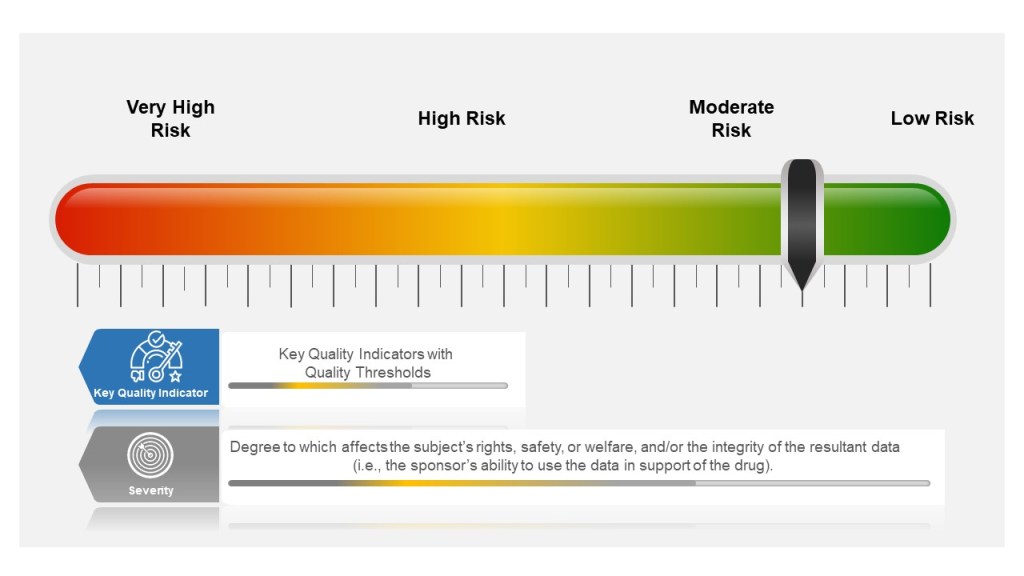
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
Events happen beyond the Protocol. These need to be managed appropriately as well.
The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Event Classification for Clinical Protocols and GCPs
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
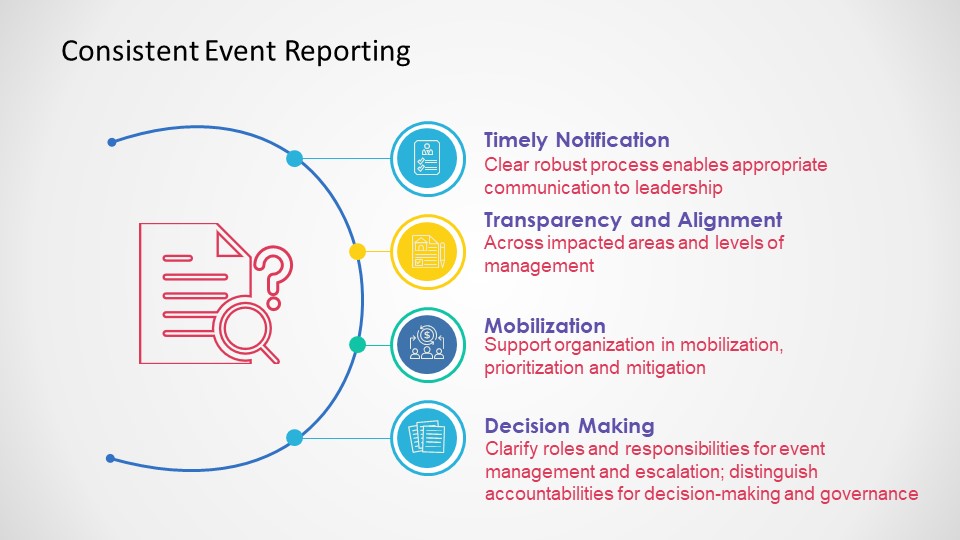
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
Events Happen
Consistent Event Reporting is Critical
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are:
Primary Investigator, Study Director, Qualified Person, Responsible Person – the pharmaceutical regulations are rife with a series of positions that are charged with achieving compliance and quality results. I tend to think of them as a giant Achilles heel created by the regulations.
The concept of an individual having all the accountability is nowhere near universal, for example, the term Quality Unit is a nice inclusive we – though I do have some quibbles on how it can end up placing the quality unit within the organization.
This is an application of the great man fallacy – the idea that one person by the brunt of education, experience, and stunning good looks can ensure product safety, efficacy and quality, and all the other aspects of patient and data integrity of trials.
That is, frankly, poppycock.
People only perform successfully when they are in a well-built system. Process drives success and leverages the right people at the right time making the right decisions with the right information. No one person can do that, and frankly thinking someone can is setting them up for failure. Which we see, a lot in the regulatory space.
Sure, the requirement exists, we need to meet it failing the agencies waking up and realizing the regulations are setting us up for failure. But we don’t need to buy into it. We build our processes to leverage the team, to democratize decisions, and to drive for reliable results.
Let’s leave the great man theory in the dustbins where it belongs.