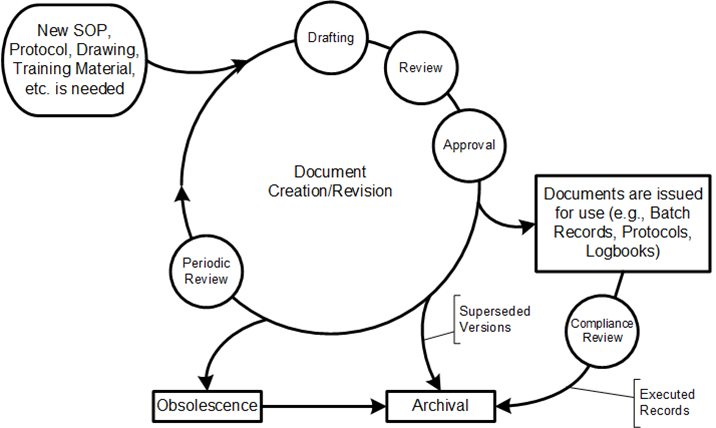
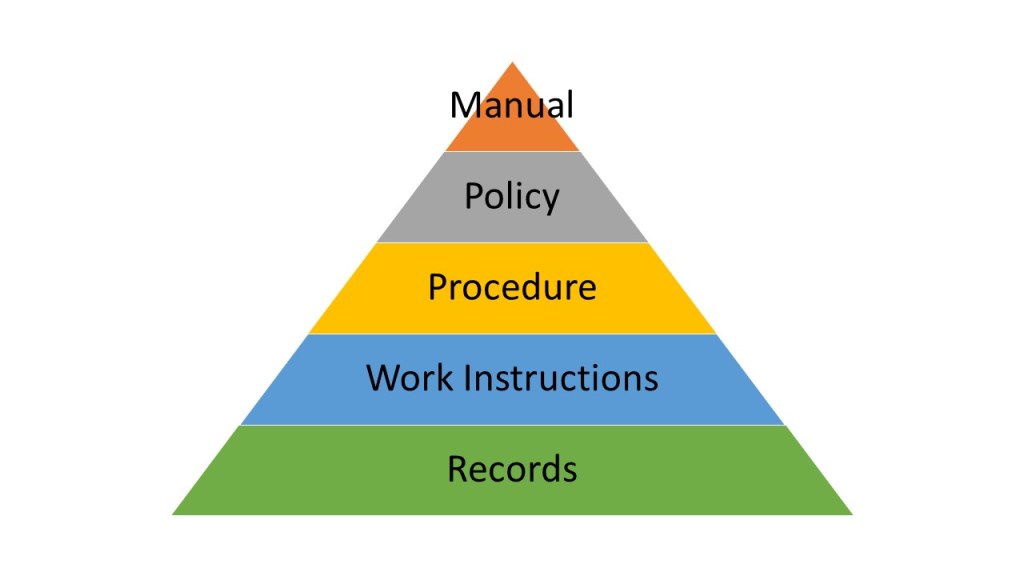
Traditional document management approaches, rooted in paper-based paradigms, create artificial boundaries between engineering activities and quality oversight. These silos become particularly problematic when implementing Quality Risk Management-based integrated Commissioning and Qualification strategies. The solution lies not in better document control procedures, but in embracing data-centric architectures that treat documents as dynamic views of underlying quality data rather than static containers of information.
The Engineering Quality Process: Beyond Document Control
The Engineering Quality Process (EQP) represents an evolution beyond traditional document management, establishing the critical interface between Good Engineering Practice and the Pharmaceutical Quality System. This integration becomes particularly crucial when we consider that engineering documents are not merely administrative artifacts—they are the embodiment of technical knowledge that directly impacts product quality and patient safety.
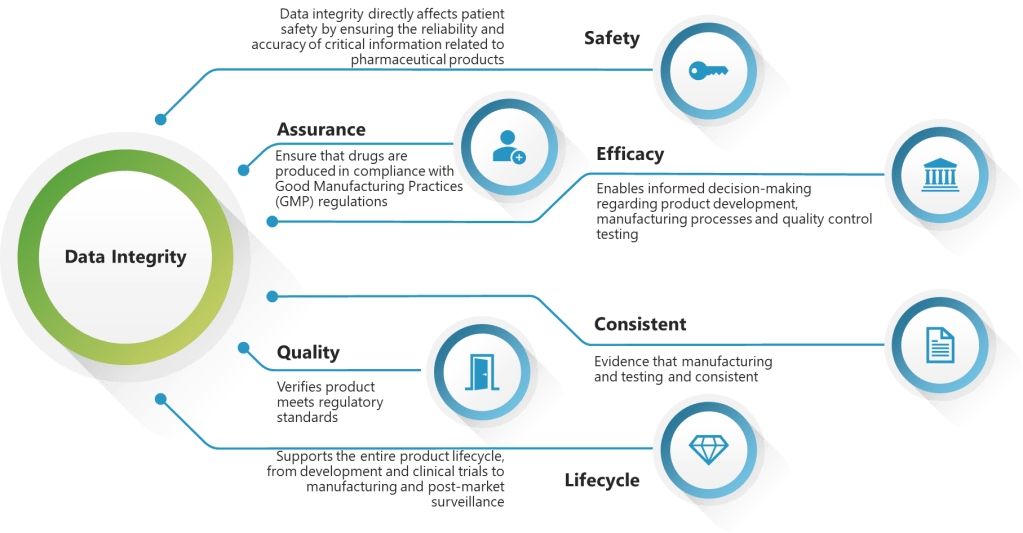
EQP implementation requires understanding that documents exist within complex data ecosystems where engineering specifications, risk assessments, change records, and validation protocols are interconnected through multiple quality processes. The challenge lies in creating systems that maintain this connectivity while ensuring ALCOA+ principles are embedded throughout the document lifecycle.
Building Systematic Document Governance
The foundation of effective GEP document management begins with recognizing that documents serve multiple masters—engineering teams need technical accuracy and accessibility, quality assurance requires compliance and traceability, and operations demands practical usability. This multiplicity of requirements necessitates what I call “multi-dimensional document governance”—systems that can simultaneously satisfy engineering, quality, and operational needs without creating redundant or conflicting documentation streams.
Effective governance structures must establish clear boundaries between engineering autonomy and quality oversight while ensuring seamless information flow across these interfaces. This requires moving beyond simple approval workflows toward sophisticated quality risk management integration where document criticality drives the level of oversight and control applied.
Electronic Quality Management System Integration: The Technical Architecture
The integration of eQMS platforms with engineering documentation can be surprisingly complex. The fundamental issue is that most eQMS solutions were designed around quality department workflows, while engineering documents flow through fundamentally different processes that emphasize technical iteration, collaborative development, and evolutionary refinement.
Core Integration Principles
Unified Data Models: Rather than treating engineering documents as separate entities, leading implementations create unified data models where engineering specifications, quality requirements, and validation protocols share common data structures. This approach eliminates the traditional handoffs between systems and creates seamless information flow from initial design through validation and into operational maintenance.
Risk-Driven Document Classification: We need to move beyond user driven classification and implement risk classification algorithms that automatically determine the level of quality oversight required based on document content, intended use, and potential impact on product quality. This automated classification reduces administrative burden while ensuring critical documents receive appropriate attention.
Contextual Access Controls: Advanced eQMS platforms provide dynamic permission systems that adjust access rights based on document lifecycle stage, user role, and current quality status. During active engineering development, technical teams have broader access rights, but as documents approach finalization and quality approval, access becomes more controlled and audited.
Validation Management System Integration
The integration of electronic Validation Management Systems (eVMS) represents a particularly sophisticated challenge because validation activities span the boundary between engineering development and quality assurance. Modern implementations create bidirectional data flows where engineering documents automatically populate validation protocols, while validation results feed back into engineering documentation and quality risk assessments.
Protocol Generation: Advanced systems can automatically generate validation protocols from engineering specifications, user requirements, and risk assessments. This automation ensures consistency between design intent and validation activities while reducing the manual effort typically required for protocol development.
Evidence Linking: Sophisticated eVMS platforms create automated linkages between engineering documents, validation protocols, execution records, and final reports. These linkages ensure complete traceability from initial requirements through final qualification while maintaining the data integrity principles essential for regulatory compliance.
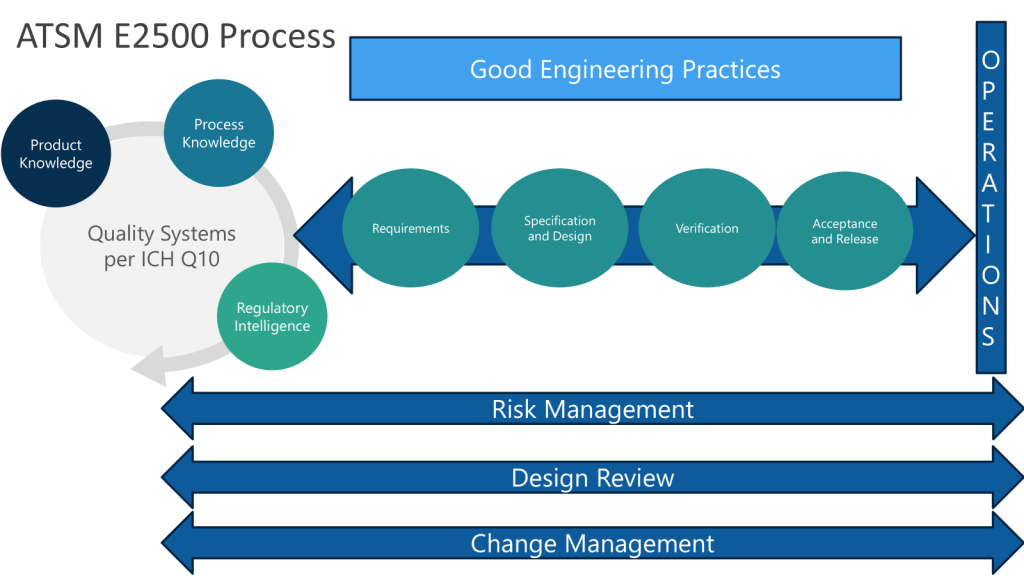
Continuous Verification: Modern systems support continuous verification approaches aligned with ASTM E2500 principles, where validation becomes an ongoing process integrated with change management rather than discrete qualification events.
Data Integrity Foundations: ALCOA+ in Engineering Documentation
The application of ALCOA+ principles to engineering documentation can create challenges because engineering processes involve significant collaboration, iteration, and refinement—activities that can conflict with traditional interpretations of data integrity requirements. The solution lies in understanding that ALCOA+ principles must be applied contextually, with different requirements during active development versus finalized documentation.
Attributability in Collaborative Engineering
Engineering documents often represent collective intelligence rather than individual contributions. Address this challenge through granular attribution mechanisms that can track individual contributions to collaborative documents while maintaining overall document integrity. This includes sophisticated version control systems that maintain complete histories of who contributed what content, when changes were made, and why modifications were implemented.
Contemporaneous Recording in Design Evolution
Traditional interpretations of contemporaneous recording can conflict with engineering design processes that involve iterative refinement and retrospective analysis. Implement design evolution tracking that captures the timing and reasoning behind design decisions while allowing for the natural iteration cycles inherent in engineering development.
Managing Original Records in Digital Environments
The concept of “original” records becomes complex in engineering environments where documents evolve through multiple versions and iterations. Establish authoritative record concepts where the system maintains clear designation of authoritative versions while preserving complete historical records of all iterations and the reasoning behind changes.
Best Practices for eQMS Integration
Systematic Architecture Design
Effective eQMS integration begins with architectural thinking rather than tool selection. Organizations must first establish clear data models that define how engineering information flows through their quality ecosystem. This includes mapping the relationships between user requirements, functional specifications, design documents, risk assessments, validation protocols, and operational procedures.
Cross-Functional Integration Teams: Successful implementations establish integrated teams that include engineering, quality, IT, and operations representatives from project inception. These teams ensure that system design serves all stakeholders’ needs rather than optimizing for a single department’s workflows.
Phased Implementation Strategies: Rather than attempting wholesale system replacement, leading organizations implement phased approaches that gradually integrate engineering documentation with quality systems. This allows for learning and refinement while maintaining operational continuity.
Change Management Integration
The integration of change management across engineering and quality systems represents a critical success factor. Create unified change control processes where engineering changes automatically trigger appropriate quality assessments, risk evaluations, and validation impact analyses.
Automated Impact Assessment: Ensure your system can automatically assess the impact of engineering changes on existing validation status, quality risk profiles, and operational procedures. This automation ensures that changes are comprehensively evaluated while reducing the administrative burden on technical teams.
Stakeholder Notification Systems: Provide contextual notifications to relevant stakeholders based on change impact analysis. This ensures that quality, operations, and regulatory affairs teams are informed of changes that could affect their areas of responsibility.
Knowledge Management Integration
Capturing Engineering Intelligence
One of the most significant opportunities in modern GEP document management lies in systematically capturing engineering intelligence that traditionally exists only in informal networks and individual expertise. Implement knowledge harvesting mechanisms that can extract insights from engineering documents, design decisions, and problem-solving approaches.
Design Decision Rationale: Require and capture the reasoning behind engineering decisions, not just the decisions themselves. This creates valuable organizational knowledge that can inform future projects while providing the transparency required for quality oversight.
Lessons Learned Integration: Rather than maintaining separate lessons learned databases, integrate insights directly into engineering templates and standard documents. This ensures that organizational knowledge is immediately available to teams working on similar challenges.
Expert Knowledge Networks
Create dynamic expert networks where subject matter experts are automatically identified and connected based on document contributions, problem-solving history, and technical expertise areas. These networks facilitate knowledge transfer while ensuring that critical engineering knowledge doesn’t remain locked in individual experts’ experience.
Technology Platform Considerations
System Architecture Requirements
Effective GEP document management requires platform architectures that can support complex data relationships, sophisticated workflow management, and seamless integration with external engineering tools. This includes the ability to integrate with Computer-Aided Design systems, engineering calculation tools, and specialized pharmaceutical engineering software.
API Integration Capabilities: Modern implementations require robust API frameworks that enable integration with the diverse tool ecosystem typically used in pharmaceutical engineering. This includes everything from CAD systems to process simulation software to specialized validation tools.
Scalability Considerations: Pharmaceutical engineering projects can generate massive amounts of documentation, particularly during complex facility builds or major system implementations. Platforms must be designed to handle this scale while maintaining performance and usability.
Validation and Compliance Framework
The platforms supporting GEP document management must themselves be validated according to pharmaceutical industry standards. This creates unique challenges because engineering systems often require more flexibility than traditional quality management applications.
GAMP 5 Compliance: Follow GAMP 5 principles for computerized system validation while maintaining the flexibility required for engineering applications. This includes risk-based validation approaches that focus validation efforts on critical system functions.
Continuous Compliance: Modern systems support continuous compliance monitoring rather than point-in-time validation. This is particularly important for engineering systems that may receive frequent updates to support evolving project needs.
Building Organizational Maturity
Cultural Transformation Requirements
The successful implementation of integrated GEP document management requires cultural transformation that goes beyond technology deployment. Engineering organizations must embrace quality oversight as value-adding rather than bureaucratic, while quality organizations must understand and support the iterative nature of engineering development.
Cross-Functional Competency Development: Success requires developing transdisciplinary competence where engineering professionals understand quality requirements and quality professionals understand engineering processes. This shared understanding is essential for creating systems that serve both communities effectively.
Evidence-Based Decision Making: Organizations must cultivate cultures that value systematic evidence gathering and rigorous analysis across both technical and quality domains. This includes establishing standards for what constitutes adequate evidence for engineering decisions and quality assessments.
Maturity Model Implementation
Organizations can assess and develop their GEP document management capabilities using maturity model frameworks that provide clear progression paths from reactive document control to sophisticated knowledge-enabled quality systems.
Level 1 – Reactive: Basic document control with manual processes and limited integration between engineering and quality systems.
Level 2 – Developing: Electronic systems with basic workflow automation and beginning integration between engineering and quality processes.
Level 3 – Systematic: Comprehensive eQMS integration with risk-based document management and sophisticated workflow automation.
Level 4 – Integrated: Unified data architectures with seamless information flow between engineering, quality, and operational systems.
Level 5 – Optimizing: Knowledge-enabled systems with predictive analytics, automated intelligence extraction, and continuous improvement capabilities.
Future Directions and Emerging Technologies
Artificial Intelligence Integration
The convergence of AI technologies with GEP document management creates unprecedented opportunities for intelligent document analysis, automated compliance checking, and predictive quality insights. The promise is systems that can analyze engineering documents to identify potential quality risks, suggest appropriate validation strategies, and automatically generate compliance reports.
Natural Language Processing: AI-powered systems can analyze technical documents to extract key information, identify inconsistencies, and suggest improvements based on organizational knowledge and industry best practices.
Predictive Analytics: Advanced analytics can identify patterns in engineering decisions and their outcomes, providing insights that improve future project planning and risk management.
Building Excellence Through Integration
The transformation of GEP document management from compliance-driven bureaucracy to value-creating knowledge systems represents one of the most significant opportunities available to pharmaceutical organizations. Success requires moving beyond traditional document control paradigms toward data-centric architectures that treat documents as dynamic views of underlying quality data.
The integration of eQMS platforms with engineering workflows, when properly implemented, creates seamless quality ecosystems where engineering intelligence flows naturally through validation processes and into operational excellence. This integration eliminates the traditional handoffs and translation losses that have historically plagued pharmaceutical quality systems while maintaining the oversight and control required for regulatory compliance.
Organizations that embrace these integrated approaches will find themselves better positioned to implement Quality by Design principles, respond effectively to regulatory expectations for science-based quality systems, and build the organizational knowledge capabilities required for sustained competitive advantage in an increasingly complex regulatory environment.
The future belongs to organizations that can seamlessly blend engineering excellence with quality rigor through sophisticated information architectures that serve both engineering creativity and quality assurance requirements. The technology exists; the regulatory framework supports it; the question remaining is organizational commitment to the cultural and architectural transformations required for success.
As we continue evolving toward more evidence-based quality practice, the organizations that invest in building coherent, integrated document management systems will find themselves uniquely positioned to navigate the increasing complexity of pharmaceutical quality requirements while maintaining the engineering innovation essential for bringing life-saving products to market efficiently and safely.