A task is the steps for doing a particular piece of work.
Procedure are activities made up of a series of tasks.
A process is an upper level description of a series of activities required to accomplish an objective. Processes are made up of procedures or tasks. They have inputs and outputs.
Process
Procedure
Task
Flow of sequences of activities that transform input elements into results
Specific and required way to carry out a process
Describe the correct steps to perform a specific task
What we do By Whom Where it takes place When it happens
How the work must be performed
How to accomplish a specific task within a process with very detailed directions
Orchestration the work
Mandatory method
Mandatory guidance
Can link to 0, 1 or more procedures
It may consist of 0, 1 or more task instructions
Focus on the instructions of 1 task
Transversal by business units
Cross functional or only 1 business unit
Only 1 business unit
Participate more than one role
Participate more than one role
Participate only one role
Encapsulates activities
Explains how to do but doesn’t get to all the details of how it is done
All of the detail of all the steps to follow in an activity
Provides the workflow model at the highest level using BPMN
Document with both narrative and images, usually in the form of use cases and workflow diagrams
Document with the maximum detail that explains step by step the instructions that must be carried out in an activity
It is pretty standard advice that relevant references to other documents should be listed in a separate section of the procedure. The reasoning is that when some standard operating procedures are intimately linked to others – the information contained in more than one document is necessary to complete a task – it is useful to include a cross-reference section in each document. Many also say that this section reinforces the SOP’s authority.
Another fairly common piece of advice is to have this, or another, section in the procedure identify the documents used in the development of the procedure, such as regulatory documents or technical/validation reports.
My take is that neither belongs in a process/procedure (SOP/WI). We should be looking to streamline requirements documents, and these sections are just cruft.
If you have electronic document control systems then cross-references should be handled trough hyperlink. Users are quite comfortable with hyperlinks and will easily navigate between documents.
Listing of regulations and other requirements belongs in a separate design document (ideally part of the document control system), and again add little value to the execution of the document.
There are a lot of so-called “best practices” about documents that stem from the days where everything is paper, and it is okay to move beyond them.
The Process Owner defines the process, including people, process steps, and technology, as well as the connections to other processes. They are accountable for change management, training, monitoring and control of the process and supporting procedure. The Process Owners owns the continuous improvement of the overall process.
Quality is ultimately responsible for the decisions made and that they align, at a minimum, with all regulatory requirements and internal standards.
Functional Area Management represents the areas that have responsibilities in the process and has a vested interest or concern in the ongoing performance of a process. This can include stakeholders who are process owners in upstream or downstream processes.
A Subject Matter Expert (SME) is typically an expert on a narrow division of a process, such as a specific tool, system, or set of process steps. A process may have multiple subject matter experts associated with it, each with varying degrees of understanding of the over-arching process.
A Risk Based Approach
The level of review of a new or revised process/procedure is guided by three fundamental risk questions:
What might go wrong with the associated process? (risk identification)
What is the likelihood that this will go wrong? (risk analysis)
What are the consequences? How severe are they if this goes wrong? (risk analysis)
Conducting risk identification is real about understanding how complicated and complex the associated process is. This looks at the following criteria:
Interconnectedness: the organization and interaction of system components and other processes
Repeatability: the amount of variance in the process
Information content: the amount of information needed to interact with the process
What Happens During a Review of Process and Procedure
The review of a process/procedure ensures that the proposed changes add value to the process and attain the outcome the organization wants. There are three levels of review (which can and often do happen simultaneously):
Functional review
Expert review by subject matter experts
Step-by-step real-world challenge
Functional review is the vetting of the process/procedure. Process stakeholders, including functional area management affected by the change has the opportunity to review the draft, suggest changes and agree to move forward.
Functional review supplies the lowest degree of assurance. This review looks for potential impact of the change on the function – usually focused on responsibilities – but does not necessarily assures a critical review.
In the case of expert review, the SMEs will review the draft for both positive and negative elements. On the positive side, they will look for the best practices, value-adding steps, flexibility in light of changing demands, scalability in light of changing output targets, etc. On the negative side, they will look for bottlenecks in the process, duplication of effort, unnecessary tasks, non-value-adding steps, role ambiguities (i.e. several positions responsible for a task, or no one responsible for a task), etc.
Expert review provides a higher degree of assurance because it is a compilation of expert opinion and it is focused on the technical content of the procedure.
The real-world challenge tests the process/procedure’s applicability by challenging it step-by-step in as much as possible the actual conditions of use. Tis involves selecting seasoned employee(s) within the scope of the draft procedure – not necessarily a SME – and comparing the steps as drafted with the actual activities. It is important to ascertain if they align. It is equally important to consider evidence of resistance, repetition and human factor problems.
Sometimes it can be more appropriate to do the real-world test as a tabletop or simulation exercise.
As sufficient reviews are obtained, the comments received are incorporated, as appropriate. Significant changes incorporated during the review process may require the procedure be re-routed for review, and may require the need to add additional reviews.
Repeat as a iterative process as necessary.
Design lifecycle
The process/procedure lifecycle can be seen as the iterative design lifecycle.
Design Thinking: Determine process needs.
Collect and document business requirements
Map current-state processes.
Observe and interview process workers.
Design process to-be.
Startup: Create process documentation, workflows, and support materials. Review and described above
Transparency is something that regulatory agencies need to get better at, both in sharing more and doing it in a timely manner. The fact that the 2019 data from the MHRA was released in October of 2020 is pretty poor. As a reference, the FDA releases their data pretty reliably at the end of the calendar year for the given year.
Been evaluating the MHRA’s 2020 data on Chapter 4 Documentation, which is the 2nd largest category of observations in 2019 (and in 2018 before it).
80 different inspections cited comments against the Principles section
Good documentation constitutes an essential part of the quality assurance system and is key to operating in compliance with GMP requirements. The various types of documents and media used should be fully defined in the manufacturer’s Quality Management System.
Documentation may exist in a variety of forms, including paper-based, electronic or photographic media. The main objective of the system of documentation utilized must be to establish, control, monitor and record all activities which directly or indirectly impact on all aspects of the quality of medicinal products. The Quality Management System should include sufficient instructional detail to facilitate a common understanding of the requirements, in addition to providing for sufficient recording of the various processes and evaluation of any observations, so that ongoing application of the requirements may be demonstrated.
There are two primary types of documentation used to manage and record GMP compliance: instructions (directions, requirements) and records/reports. Appropriate good documentation practice should be applied with respect to the type of document.
Suitable controls should be implemented to ensure the accuracy, integrity, availability and legibility of documents. Instruction documents should be free from errors and available in writing. The term ‘written’ means recorded, or documented on media from which data may be rendered in a human readable form.
Principles, Chapter 4 Documentation
The Principles section then goes on to lay out the required document types.
We can learn more by drilling down in the document.
There are 87 inspections with 4.1 in section “Generation and Control of Documents”. 1 is critical and 25 are major. Here we see failures in understanding types of documents and controlling them, or maybe just having them in the first place.
The 82 against 4.2 (1 critical and 20 major) are more about having the manufacturing and testing process defined (and matching the filing).
103 inspections with observations against 4.3 (23 major) show companies that do not have appropriate approval and release controls
14 for 4.4 (6 major) means there are 14 companies out there who can’t write a good process and procedure. 4.4 has one of my favorite requirements “written in an imperative mandatory style”
60 against 4.5 (13 major) demonstrates a lack of review and keeping documents up-to-date.
12 companies (6 major) have terrible handwriting and cannot stick to ballpoints, yes in fact 4.7 states “Handwritten entries should be made in clear, legible, indelible way.”
We write and use procedures to help the user complete the task successfully and avoid undesired outcomes. Well-written procedures are an integral part of any organization for operation, managing risks, and continuous improvement. Effective procedures are important for the transfer of knowledge from the engineers/architects of the system to the users of the system.
Good procedures, and we are not talking format so this can be paper documents to a mixed reality guide, provide these four categories of information:
Goal: The goal presented to the user as a state to be realized. This can be an end state or an intermediate state of the overall system.
Prerequisites: The condition for moving toward the desired state or goal. These are the conditions that must be satisfied so that the user can achieve the goal.
Actions and reactions: These states are reached through actions of the user and the reactions of the system. They may have milestones or sub-goals. It involves the description of (a series of) action steps.
Unwanted: These are the states to be avoided (e.g., errors, malfunctions, injuries). It provides guidelines on what to avoid for successful and safe execution of procedure and may include warning, caution, or instruction for solving a potential problem.
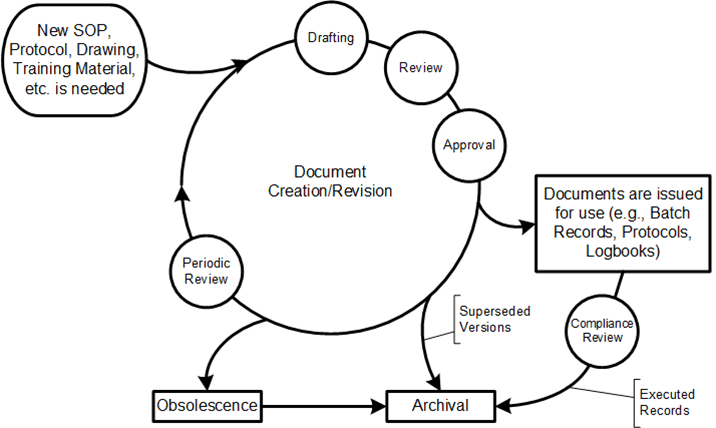
Procedures have a lifecycle through which they are developed, administered, used, reviewed, and updated. In the post “Document Management” I discussed the document management lifecycle.
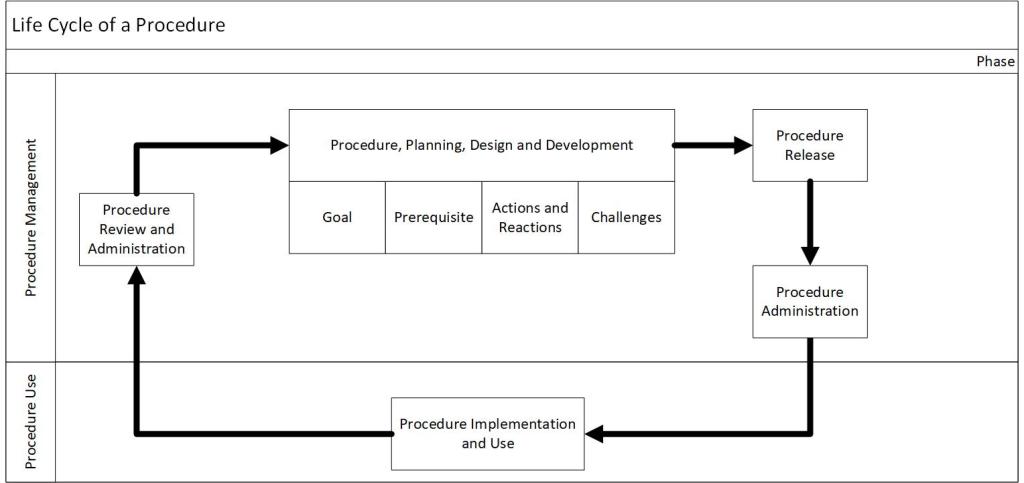
I want to focus specifically on procedures by covering five distinct phases: procedure plan, design and development, procedure authorization, procedure administration, procedure implementation and use, and procedure review and maintenance.
Identifying whether a procedure is necessary; collecting required information; producing instructions and information on the work, regulatory compliance, process and personnel safety; a walkthrough to ensure quality and potential compliance of the procedure
“New SOP is needed” Drafting
Procedure Authorization
Procedure review; publishing the final document; revision control; the approval process.