Over the past decades, as I’ve grown and now led quality organizations in biotechnology, I’ve encountered many thinkers who’ve shaped my approach to investigation and risk management. But few have fundamentally altered my perspective like Sidney Dekker. His work didn’t just add to my toolkit—it forced me to question some of my most basic assumptions about human error, system failure, and what it means to create genuinely effective quality systems.
Dekker’s challenge to move beyond “safety theater” toward authentic learning resonates deeply with my own frustrations about quality systems that look impressive on paper but fail when tested by real-world complexity.
Why Dekker Matters for Quality Leaders
Professor Sidney Dekker brings a unique combination of academic rigor and operational experience to safety science. As both a commercial airline pilot and the Director of the Safety Science Innovation Lab at Griffith University, he understands the gap between how work is supposed to happen and how it actually gets done. This dual perspective—practitioner and scholar—gives his critiques of traditional safety approaches unusual credibility.
But what initially drew me to Dekker’s work wasn’t his credentials. It was his ability to articulate something I’d been experiencing but couldn’t quite name: the growing disconnect between our increasingly sophisticated compliance systems and our actual ability to prevent quality problems. His concept of “drift into failure” provided a framework for understanding why organizations with excellent procedures and well-trained personnel still experience systemic breakdowns.
The “New View” Revolution
Dekker’s most fundamental contribution is what he calls the “new view” of human error—a complete reframing of how we understand system failures. Having spent years investigating deviations and CAPAs, I can attest to how transformative this shift in perspective can be.
The Traditional Approach I Used to Take:
Human error causes problems
People are unreliable; systems need protection from human variability
Solutions focus on better training, clearer procedures, more controls
Dekker’s New View That Changed My Practice:
Human error is a symptom of deeper systemic issues
People are the primary source of system reliability, not the threat to it
Variability and adaptation are what make complex systems work
This isn’t just academic theory—it has practical implications for every investigation I lead. When I encounter “operator error” in a deviation investigation, Dekker’s framework pushes me to ask different questions: What made this action reasonable to the operator at the time? What system conditions shaped their decision-making? How did our procedures and training actually perform under real-world conditions?
This shift aligns perfectly with the causal reasoning approaches I’ve been developing on this blog. Instead of stopping at “failure to follow procedure,” we dig into the specific mechanisms that drove the event—exactly what Dekker’s view demands.
Drift Into Failure: Why Good Organizations Go Bad
Perhaps Dekker’s most powerful concept for quality leaders is “drift into failure”—the idea that organizations gradually migrate toward disaster through seemingly rational local decisions. This isn’t sudden catastrophic failure; it’s incremental erosion of safety margins through competitive pressure, resource constraints, and normalized deviance.
I’ve seen this pattern repeatedly. For example, a cleaning validation program starts with robust protocols, but over time, small shortcuts accumulate: sampling points that are “difficult to access” get moved, hold times get shortened when production pressure increases, acceptance criteria get “clarified” in ways that gradually expand limits.
Each individual decision seems reasonable in isolation. But collectively, they represent drift—a gradual migration away from the original safety margins toward conditions that enable failure. The contamination events and data integrity issues that plague our industry often represent the endpoint of these drift processes, not sudden breakdowns in otherwise reliable systems.
Traditional root cause analysis seeks the single factor that “caused” an event, but complex system failures emerge from multiple interacting conditions. The take-the-best heuristic I’ve been exploring on this blog—focusing on the most causally powerful factor—builds directly on Dekker’s insight that we need to understand mechanisms, not hunt for someone to blame.
When I investigate a failure now, I’m not looking for THE root cause. I’m trying to understand how various factors combined to create conditions for failure. What pressures were operators experiencing? How did procedures perform under actual conditions? What information was available to decision-makers? What made their actions reasonable given their understanding of the situation?
This approach generates investigations that actually help prevent recurrence rather than just satisfying regulatory expectations for “complete” investigations.
Just Culture: Moving Beyond Blame
Dekker’s evolution of just culture thinking has been particularly influential in my leadership approach. His latest work moves beyond simple “blame-free” environments toward restorative justice principles—asking not “who broke the rule” but “who was hurt and how can we address underlying needs.”
This shift has practical implications for how I handle deviations and quality events. Instead of focusing on disciplinary action, I’m asking: What systemic conditions contributed to this outcome? What support do people need to succeed? How can we address the underlying vulnerabilities this event revealed?
This doesn’t mean eliminating accountability—it means creating accountability systems that actually improve performance rather than just satisfying our need to assign blame.
Safety Theater: The Problem with Compliance Performance
Dekker’s most recent work on “safety theater” hits particularly close to home in our regulated environment. He defines safety theater as the performance of compliance when under surveillance that retreats to actual work practices when supervision disappears.
I’ve watched organizations prepare for inspections by creating impressive documentation packages that bear little resemblance to how work actually gets done. Procedures get rewritten to sound more rigorous, training records get updated, and everyone rehearses the “right” answers for auditors. But once the inspection ends, work reverts to the adaptive practices that actually make operations function.
This theater emerges from our desire for perfect, controllable systems, but it paradoxically undermines genuine safety by creating inauthenticity. People learn to perform compliance rather than create genuine safety and quality outcomes.
The falsifiable quality systems I’ve been advocating on this blog represent one response to this problem—creating systems that can be tested and potentially proven wrong rather than just demonstrated as compliant.
Six Practical Takeaways for Quality Leaders
After years of applying Dekker’s insights in biotechnology manufacturing, here are the six most practical lessons for quality professionals:
1. Treat “Human Error” as the Beginning of Investigation, Not the End
When investigations conclude with “human error,” they’ve barely started. This should prompt deeper questions: Why did this action make sense? What system conditions shaped this decision? What can we learn about how our procedures and training actually perform under pressure?
2. Understand Work-as-Done, Not Just Work-as-Imagined
There’s always a gap between procedures (work-as-imagined) and actual practice (work-as-done). Understanding this gap and why it exists is more valuable than trying to force compliance with unrealistic procedures. Some of the most important quality improvements I’ve implemented came from understanding how operators actually solve problems under real conditions.
3. Measure Positive Capacities, Not Just Negative Events
Traditional quality metrics focus on what didn’t happen—no deviations, no complaints, no failures. I’ve started developing metrics around investigation quality, learning effectiveness, and adaptive capacity rather than just counting problems. How quickly do we identify and respond to emerging issues? How effectively do we share learning across sites? How well do our people handle unexpected situations?
4. Create Psychological Safety for Learning
Fear and punishment shut down the flow of safety-critical information. Organizations that want to learn from failures must create conditions where people can report problems, admit mistakes, and share concerns without fear of retribution. This is particularly challenging in our regulated environment, but it’s essential for moving beyond compliance theater toward genuine learning.
5. Focus on Contributing Conditions, Not Root Causes
Complex failures emerge from multiple interacting factors, not single root causes. The take-the-best approach I’ve been developing helps identify the most causally powerful factor while avoiding the trap of seeking THE cause. Understanding mechanisms is more valuable than finding someone to blame.
6. Embrace Adaptive Capacity Instead of Fighting Variability
People’s ability to adapt and respond to unexpected conditions is what makes complex systems work, not a threat to be controlled. Rather than trying to eliminate human variability through ever-more-prescriptive procedures, we should understand how that variability creates resilience and design systems that support rather than constrain adaptive problem-solving.
Connection to Investigation Excellence
Dekker’s work provides the theoretical foundation for many approaches I’ve been exploring on this blog. His emphasis on testable hypotheses rather than compliance theater directly supports falsifiable quality systems. His new view framework underlies the causal reasoning methods I’ve been developing. His focus on understanding normal work, not just failures, informs my approach to risk management.
Most importantly, his insistence on moving beyond negative reasoning (“what didn’t happen”) to positive causal statements (“what actually happened and why”) has transformed how I approach investigations. Instead of documenting failures to follow procedures, we’re understanding the specific mechanisms that drove events—and that makes all the difference in preventing recurrence.
Essential Reading for Quality Leaders
If you’re leading quality organizations in today’s complex regulatory environment, these Dekker works are essential:
Dekker’s work challenges us as quality leaders to move beyond the comfortable certainty of compliance-focused approaches toward the more demanding work of creating genuine learning systems. This requires admitting that our procedures and training might not work as intended. It means supporting people when they make mistakes rather than just punishing them. It demands that we measure our success by how well we learn and adapt, not just how well we document compliance.
This isn’t easy work. It requires the kind of organizational humility that Amy Edmondson and other leadership researchers emphasize—the willingness to be proven wrong in service of getting better. But in my experience, organizations that embrace this challenge develop more robust quality systems and, ultimately, better outcomes for patients.
The question isn’t whether Sidney Dekker is right about everything—it’s whether we’re willing to test his ideas and learn from the results. That’s exactly the kind of falsifiable approach that both his work and effective quality systems demand.
Problem-solving is too often shaped by the assumption that the system is perfectly understood and fully specified. If something goes wrong—a deviation, a batch out-of-spec, or a contamination event—our approach is to dissect what “failed” and fix that flaw, believing this will restore order. This way of thinking, which I call the malfunction mindset, is as ingrained as it is incomplete. It assumes that successful outcomes are the default, that work always happens as written in SOPs, and that only failure deserves our scrutiny.
But here’s the paradox: most of the time, our highly complex manufacturing environments actually succeed—often under imperfect, shifting, and not fully understood conditions. If we only study what failed, and never question how our systems achieve their many daily successes, we miss the real nature of pharmaceutical quality: it is not the absence of failure, but the presence of robust, adaptive work. Taking this broader, more nuanced perspective is not just an academic exercise—it’s essential for building resilient operations that truly protect patients, products, and our organizations.
Drawing from my thinking through zemblanity (the predictable but often overlooked negative outcomes of well-intentioned quality fixes), the effectiveness paradox (why “nothing bad happened” isn’t proof your quality system works), and the persistent gap between work-as-imagined and work-as-done, this post explores why the malfunction mindset persists, how it distorts investigations, and what future-ready quality management should look like.
The Allure—and Limits—of the Failure Model
Why do we reflexively look for broken parts and single points of failure? It is, as Sidney Dekker has argued, both comforting and defensible. When something goes wrong, you can always point to a failed sensor, a missed checklist, or an operator error. This approach—introducing another level of documentation, another check, another layer of review—offers a sense of closure and regulatory safety. After all, as long as you can demonstrate that you “fixed” something tangible, you’ve fulfilled investigational due diligence.
Yet this fails to account for how quality is actually produced—or lost—in the real world. The malfunction model treats systems like complicated machines: fix the broken gear, oil the creaky hinge, and the machine runs smoothly again. But, as Dekker reminds us in Drift Into Failure, such linear thinking ignores the drift, adaptation, and emergent complexity that characterize real manufacturing environments. The truth is, in complex adaptive systems like pharmaceutical manufacturing, it often takes more than one “error” for failure to manifest. The system absorbs small deviations continuously, adapting and flexing until, sometimes, a boundary is crossed and a problem surfaces.
W. Edwards Deming’s wisdom rings truer than ever: “Most problems result from the system itself, not from individual faults.” A sustainable approach to quality is one that designs for success—and that means understanding the system-wide properties enabling robust performance, not just eliminating isolated malfunctions.
Procedural Fundamentalism: The Work-as-Imagined Trap
One of the least examined, yet most impactful, contributors to the malfunction mindset is procedural fundamentalism—the belief that the written procedure is both a complete specification and an accurate description of work. This feels rigorous and provides compliance comfort, but it is a profound misreading of how work actually happens in pharmaceutical manufacturing.
Work-as-imagined, as elucidated by Erik Hollnagel and others, represents an abstraction: it is how distant architects of SOPs visualize the “correct” execution of a process. Yet, real-world conditions—resource shortages, unexpected interruptions, mismatched raw materials, shifting priorities—force adaptation. Operators, supervisors, and Quality professionals do not simply “follow the recipe”: they interpret, improvise, and—crucially—adjust on the fly.
When we treat procedures as authoritative descriptions of reality, we create the proxy problem: our investigations compare real operations against an imagined baseline that never fully existed. Deviations become automatically framed as problem points, and success is redefined as rigid adherence, regardless of context or outcome.
Complexity, Performance Variability, and Real Success
So, how do pharmaceutical operations succeed so reliably despite the ever-present complexity and variability of daily work?
The answer lies in embracing performance variability as a feature of robust systems, not a flaw. In high-reliability environments—from aviation to medicine to pharmaceutical manufacturing—success is routinely achieved not by demanding strict compliance, but by cultivating adaptive capacity.
Consider environmental monitoring in a sterile suite: The procedure may specify precise times and locations, but a seasoned operator, noticing shifts in people flow or equipment usage, might proactively sample a high-risk area more frequently. This adaptation—not captured in work-as-imagined—actually strengthens data integrity. Yet, traditional metrics would treat this as a procedural deviation.
This is the paradox of the malfunction mindset: in seeking to eliminate all performance variability, we risk undermining precisely those adaptive behaviors that produce reliable quality under uncertainty.
Why the Malfunction Mindset Persists: Cognitive Comfort and Regulatory Reinforcement
Why do organizations continue to privilege the malfunction mindset, even as evidence accumulates of its limits? The answer is both psychological and cultural.
Component breakdown thinking is psychologically satisfying—it offers a clear problem, a specific cause, and a direct fix. For regulatory agencies, it is easy to measure and audit: did the deviation investigation determine the root cause, did the CAPA address it, does the documentation support this narrative? Anything that doesn’t fit this model is hard to defend in audits or inspections.
Yet this approach offers, at best, a partial diagnosis and, at worst, the illusion of control. It encourages organizations to catalog deviations while blindly accepting a much broader universe of unexamined daily adaptations that actually determine system robustness.
Complexity Science and the Art of Organizational Success
To move toward a more accurate—and ultimately more effective—model of quality, pharmaceutical leaders must integrate the insights of complexity science. Drawing from the work of Stuart Kauffman and others at the Santa Fe Institute, we understand that the highest-performing systems operate not at the edge of rigid order, but at the “edge of chaos,” where structure is balanced with adaptability.
In these systems, success and failure both arise from emergent properties—the patterns of interaction between people, procedures, equipment, and environment. The most meaningful interventions, therefore, address how the parts interact, not just how each part functions in isolation.
This explains why traditional root cause analysis, focused on the parts, often fails to produce lasting improvements; it cannot account for outcomes that emerge only from the collective dynamics of the system as a whole.
Investigating for Learning: The Take-the-Best Heuristic
A key innovation needed in pharmaceutical investigations is a shift to what Hollnagel calls Safety-II thinking: focusing on how things go right as well as why they occasionally go wrong.
Here, the take-the-best heuristic becomes crucial. Instead of compiling lists of all deviations, ask: Among all contributing factors, which one, if addressed, would have the most powerful positive impact on future outcomes, while preserving adaptive capacity? This approach ensures investigations generate actionable, meaningful learning, rather than feeding the endless paper chase of “compliance theater.”
Building Systems That Support Adaptive Capability
Taking complexity and adaptive performance seriously requires practical changes to how we design procedures, train, oversee, and measure quality.
Procedure Design: Make explicit the distinction between objectives and methods. Procedures should articulate clear quality goals, specify necessary constraints, but deliberately enable workers to choose methods within those boundaries when faced with new conditions.
Training: Move beyond procedural compliance. Develop adaptive expertise in your staff, so they can interpret and adjust sensibly—understanding not just “what” to do, but “why” it matters in the bigger system.
Oversight and Monitoring: Audit for adaptive capacity. Don’t just track “compliance” but also whether workers have the resources and knowledge to adapt safely and intelligently. Positive performance variability (smart adaptations) should be recognized and studied.
Quality System Design: Build systematic learning from both success and failure. Examine ordinary operations to discern how adaptive mechanisms work, and protect these capabilities rather than squashing them in the name of “control.”
Leadership and Systems Thinking
Realizing this vision depends on a transformation in leadership mindset—from one seeking control to one enabling adaptive capacity. Deming’s profound knowledge and the principles of complexity leadership remind us that what matters is not enforcing ever-stricter compliance, but cultivating an organizational context where smart adaptation and genuine learning become standard.
Leadership must:
Distinguish between complicated and complex: Apply detailed procedures to the former (e.g., calibration), but support flexible, principles-based management for the latter.
Tolerate appropriate uncertainty: Not every problem has a clear, single answer. Creating psychological safety is essential for learning and adaptation during ambiguity.
Develop learning organizations: Invest in deep understanding of operations, foster regular study of work-as-done, and celebrate insights from both expected and unexpected sources.
Practical Strategies for Implementation
Turning these insights into institutional practice involves a systematic, research-inspired approach:
Start procedure development with observation of real work before specifying methods. Small scale and mock exercises are critical.
Employ cognitive apprenticeship models in training, so that experience, reasoning under uncertainty, and systems thinking become core competencies.
Begin investigations with appreciative inquiry—map out how the system usually works, not just how it trips up.
Measure leading indicators (capacity, information flow, adaptability) not just lagging ones (failures, deviations).
Create closed feedback loops for corrective actions—insisting every intervention be evaluated for impact on both compliance and adaptive capacity.
Scientific Quality Management and Adaptive Systems: No Contradiction
The tension between rigorous scientific quality management (QbD, process validation, risk management frameworks) and support for adaptation is a false dilemma. Indeed, genuine scientific quality management starts with humility: the recognition that our understanding of complex systems is always partial, our controls imperfect, and our frameworks provisional.
A falsifiable quality framework embeds learning and adaptation at its core—treating deviations as opportunities to test and refine models, rather than simply checkboxes to complete.
The best organizations are not those that experience the fewest deviations, but those that learn fastest from both expected and unexpected events, and apply this knowledge to strengthen both system structure and adaptive capacity.
Embracing Normal Work: Closing the Gap
Normal pharmaceutical manufacturing is not the story of perfect procedural compliance; it’s the story of people, working together to achieve quality goals under diverse, unpredictable, and evolving conditions. This is both more challenging—and more rewarding—than any plan prescribed solely by SOPs.
To truly move the needle on pharmaceutical quality, organizations must:
Embrace performance variability as evidence of adaptive capacity, not just risk.
Investigate for learning, not blame; study success, not just failure.
Design systems to support both structure and flexible adaptation—never sacrificing one entirely for the other.
Cultivate leadership that values humility, systems thinking, and experimental learning, creating a culture comfortable with complexity.
This approach will not be easy. It means questioning decades of compliance custom, organizational habit, and intellectual ease. But the payoff is immense: more resilient operations, fewer catastrophic surprises, and, above all, improved safety and efficacy for the patients who depend on our products.
The challenge—and the opportunity—facing pharmaceutical quality management is to evolve beyond compliance theater and malfunction thinking into a new era of resilience and organizational learning. Success lies not in the illusory comfort of perfectly executed procedures, but in the everyday adaptations, intelligent improvisation, and system-level capabilities that make those successes possible.
The call to action is clear: Investigate not just to explain what failed, but to understand how, and why, things so often go right. Protect, nurture, and enhance the adaptive capacities of your organization. In doing so, pharmaceutical quality can finally become more than an after-the-fact audit; it will become the creative, resilient capability that patients, regulators, and organizations genuinely want to hire.
The Catalent Indiana 483 form from July 2025 reads like a textbook example of my newest word, zemblanity, in risk management—the patterned, preventable misfortune that accrues not from blind chance, but from human agency and organizational design choices that quietly hardwire failure into our operations.
Twenty hair contamination deviations. Seven months to notify suppliers. Critical equipment failures dismissed as “not impacting SISPQ.” Media fill programs missing the very interventions they should validate. This isn’t random bad luck—it’s a quality system that has systematically normalized exactly the kinds of deviations that create inspection findings.
The Architecture of Inevitable Failure
Reading through the six major observations, three systemic patterns emerge that align perfectly with the hidden architecture of failure I discussed in my recent post on zemblanity.
Pattern 1: Investigation Theatre Over Causal Understanding
Observation 1 reveals what happens when investigations become compliance exercises rather than learning tools. The hair contamination trend—20 deviations spanning multiple product codes—received investigation resources proportional to internal requirement, not actual risk. As I’ve written about causal reasoning versus negative reasoning, these investigations focused on what didn’t happen rather than understanding the causal mechanisms that allowed hair to systematically enter sterile products.
The tribal knowledge around plunger seating issues exemplifies this perfectly. Operators developed informal workarounds because the formal system failed them, yet when this surfaced during an investigation, it wasn’t captured as a separate deviation worthy of systematic analysis. The investigation closed the immediate problem without addressing the systemic failure that created the conditions for operator innovation in the first place.
Pattern 2: Trend Blindness and Pattern Fragmentation
The most striking aspect of this 483 is how pattern recognition failed across multiple observations. Twenty-three work orders on critical air handling systems. Ten work orders on a single critical water system. Recurring membrane failures. Each treated as isolated maintenance issues rather than signals of systematic degradation.
This mirrors what I’ve discussed about normalization of deviance—where repeated occurrences of problems that don’t immediately cause catastrophe gradually shift our risk threshold. The work orders document a clear pattern of equipment degradation, yet each was risk-assessed as “not impacting SISPQ” without apparent consideration of cumulative or interactive effects.
Pattern 3: Control System Fragmentation
Perhaps most revealing is how different control systems operated in silos. Visual inspection systems that couldn’t detect the very defects found during manual inspection. Environmental monitoring that didn’t include the most critical surfaces. Media fills that omitted interventions documented as root causes of previous failures.
This isn’t about individual system inadequacy—it’s about what happens when quality systems evolve as collections of independent controls rather than integrated barriers designed to work together.
Solutions: From Zemblanity to Serendipity
Drawing from the approaches I’ve developed on this blog, here’s how Catalent could transform their quality system from one that breeds inevitable failure to one that creates conditions for quality serendipity:
Implement Causally Reasoned Investigations
The Energy Safety Canada white paper I discussed earlier this year offers a powerful framework for moving beyond counterfactual analysis. Instead of concluding that operators “failed to follow procedure” regarding stopper installation, investigate why the procedure was inadequate for the equipment configuration. Instead of noting that supplier notification was delayed seven months, understand the systemic factors that made immediate notification unlikely.
Practical Implementation:
Retrain investigators in causal reasoning techniques
Require investigation sponsors (area managers) to set clear expectations for causal analysis
Implement structured causal analysis tools like Cause-Consequence Analysis
Focus on what actually happened and why it made sense to people at the time
The take-the-best heuristic I recently explored offers a powerful lens for barrier analysis. Rather than implementing multiple independent controls, identify the single most causally powerful barrier that would prevent each failure type, then design supporting barriers that enhance rather than compete with the primary control.
For hair contamination specifically:
Implement direct stopper surface monitoring as the primary barrier
Design visual inspection systems specifically to detect proteinaceous particles
Create supplier qualification that includes contamination risk assessment
Establish real-time trend analysis linking supplier lots to contamination events
Establish Dynamic Trend Integration
Traditional trending treats each system in isolation—environmental monitoring trends, deviation trends, CAPA trends, maintenance trends. The Catalent 483 shows what happens when these parallel trend systems fail to converge into integrated risk assessment.
Integrated Trending Framework:
Create cross-functional trend review combining all quality data streams
Implement predictive analytics linking maintenance patterns to quality risks
Design Product Quality Reviews that explicitly correlate equipment performance with product quality data
Transform CAPA from Compliance to Learning
The recurring failures documented in this 483—repeated hair findings after CAPA implementation, continued equipment failures after “repair”—reflect what I’ve called the effectiveness paradox. Traditional CAPA focuses on thoroughness over causal accuracy.
CAPA Transformation Strategy:
Implement a proper CAPA hierarchy, prioritizing elimination and replacement over detection and mitigation
Establish effectiveness criteria before implementation, not after
Create learning-oriented CAPA reviews that ask “What did this teach us about our system?”
Link CAPA effectiveness directly to recurrence prevention rather than procedural compliance
Build Anticipatory Quality Architecture
The most sophisticated element would be creating what I call “quality serendipity”—systems that create conditions for positive surprises rather than inevitable failures. This requires moving from reactive compliance to anticipatory risk architecture.
Anticipatory Elements:
Implement supplier performance modeling that predicts contamination risk before it manifests
Create equipment degradation models that trigger quality assessment before failure
Establish operator feedback systems that capture emerging risks in real-time
Design quality reviews that explicitly seek weak signals of system stress
The Cultural Foundation
None of these technical solutions will work without addressing the cultural foundation that allowed this level of systematic failure to persist. The 483’s most telling detail isn’t any single observation—it’s the cumulative picture of an organization where quality indicators were consistently rationalized rather than interrogated.
As I’ve written about quality culture, without psychological safety and learning orientation, people won’t commit to building and supporting robust quality systems. The tribal knowledge around plunger seating, the normalization of recurring equipment failures, the seven-month delay in supplier notification—these suggest a culture where adaptation to system inadequacy became preferable to system improvement.
The path forward requires leadership that creates conditions for quality serendipity: reward pattern recognition over problem solving, celebrate early identification of weak signals, and create systems that make the right choice the easy choice.
Beyond Compliance: Building Anti-Fragile Quality
The Catalent 483 offers more than a cautionary tale—it provides a roadmap for quality transformation. Every observation represents an invitation to build quality systems that become stronger under stress rather than more brittle.
Organizations that master this transformation—moving from zemblanity-generating systems to serendipity-creating ones—will find that quality becomes not just a regulatory requirement but a competitive advantage. They’ll detect risks earlier, respond more effectively, and create the kind of operational resilience that turns disruption into opportunity.
The choice is clear: continue managing quality as a collection of independent compliance activities, or build integrated systems designed to create the conditions for sustained quality success. The Catalent case shows us what happens when we choose poorly. The frameworks exist to choose better.
What patterns of “inevitable failure” do you see in your own quality systems? How might shifting from negative reasoning to causal understanding transform your approach to investigations? Share your thoughts—this conversation about quality transformation is one we need to have across the industry.
Zemblanity is actually a pretty good word for our field. I’m going to test it out, see if it has legs.
Zemblanity in Risk Management: Turning the Mirror on Hidden System Fragility
If you’re reading this blog, you already know that risk management isn’t about tallying up hypothetical hazards and ticking regulatory boxes. But have you ever stopped to ask whether your systems are quietly hardwiring failure—almost by design? Christian Busch’s recent LSE Business Review article lands on a word for this: zemblanity—the “opposite of serendipity,” or, more pointedly, bad luck that’s neither blind nor random, but structured right into the bones of our operations.
This idea resonates powerfully with the transformations occurring in pharmaceutical quality systems—the same evolution guiding the draft revision of Eudralex Volume 4 Chapter 1. In both Busch’s analysis and regulatory trends, we’re urged to confront root causes, trace risk back to its hidden architecture, and actively dismantle the quiet routines and incentives that breed failure. This isn’t mere thought leadership; it’s a call to reexamine how our own practices may be cultivating fields of inevitable misfortune—the very zemblanity that keeps reputational harm and catastrophic events just a few triggers away.
The Zemblanity Field: Where Routine Becomes Risk
Let’s be honest: the ghosts in our machines are rarely accidents. They don’t erupt out of blue-sky randomness. They were grown in cultures that prized efficiency over resilience, chased short-term gains, and normalized critical knowledge gaps. In my blog post on normalization of deviance (see: “Why Normalization of Deviance Threatens your CAPA Logic”), I map out how subtle cues and “business as usual” thinking produce exactly these sorts of landmines.
Busch’s zemblanity—the patterned and preventable misfortune that accrues from human agency—makes for a brutal mirror. Risk managers must ask: Which of our controls are truly protective, and which merely deliver the warm glow of compliance while quietly amplifying vulnerability? If serendipity is a lucky break, zemblanity is the misstep built into the schedule, the fragility we invite by squeezing the system too hard.
From Hypotheticals to Archaeology: How to Evaluate Zemblanity
So, how does one bring zemblanity into practical risk management? It starts by shifting the focus from cataloguing theoretical events to archaeology: uncovering the layered decisions, assumptions, and interdependencies that have silently locked in failure modes.
1. Map Near Misses and Routine Workarounds
Stop treating near misses as flukes. Every recurrence is a signpost pointing to underlying zemblanity. Investigate not just what happened, but why the system allowed it in the first place. High-performing teams capture these “almost events” the way a root cause analyst mines deviations for actionable knowledge .
2. Scrutinize Margins and Slack
Where are your processes running on fumes? Organizations that cut every buffer in service of “efficiency” are constructing perfect conditions for zemblanity. Whether it’s staffing, redundancy in critical utilities, or quality reserves, scrutinize these margins. If slim tolerances have become your operating norm, you’re nurturing the zemblanity field.
3. Map Hidden Interdependencies
Borrowing from system dynamics and failure mode mapping, draw out the connections you typically overlook and the informal routes by which information or pressure travels. Build reverse timelines—starting at failure—to trace seemingly disparate weak points back to core drivers.
4. Interrogate Culture and Incentives
A robust risk culture isn’t measured by the thoroughness of your SOPs, but by whether staff feel safe raising “bad news” and questioning assumptions.
5. Audit Cost-Cutting and “Optimizations”
Lean initiatives and cost-cutting programs can easily morph from margin enhancement to zemblanity engines. Run post-implementation reviews of such changes: was resilience sacrificed for pennywise savings? If so, add these to your risk register, and reframe “efficiency” in light of the total cost of a fragile response to disruption.
6. Challenge “Never Happen Here” Assumptions
Every mature risk program needs a cadence of challenging assumptions. Run pre-mortem workshops with line staff and cross-functional teams to simulate how multi-factor failures could cascade. Spotlight scenarios previously dismissed as “impossible” and ask why. Highlight usage in quality system design.
Operationalizing Zemblanity in PQS
The Eudralex Chapter 1 draft’s movement from static compliance to dynamic, knowledge-centric risk management lines up perfectly here. Embedding zemblanity analysis is less about new tools and more about repurposing familiar practices: after-action reviews, bowtie diagrams, CAPA trend analysis, incident logs—all sharpened with explicit attention to how our actions and routines cultivate not just risk, but structural misfortune.
Your Product Quality Review (PQR) process, for instance, should now interrogate near misses, not just reject rates or OOS incidents. It is time to pivot from dull data reviews reviews to causal inference—asking how past knowledge blind spots or hasty “efficiencies” became hazards.
And as pharmaceutical supply chains grow ever more interdependent and brittle, proactive risk detection needs routine revisiting. Integrate zemblanity logic into your risk and resilience dashboards—flag not just frequency, but pattern, agency, and the cultural drivers of preventable failures.
Risk professionals can no longer limit themselves to identifying hazards and correcting defects post hoc. Proactive knowledge management and an appetite for self-interrogation will mark the difference between organizations set up for breakthroughs and those unwittingly primed for avoidable disaster.
The challenge—echoed in both Busch’s argument and the emergent GMP landscape—is clear: shrink the zemblanity field. Turn pattern-seeking into your default. Reward curiosity within your team. Build analytic vigilance into every level of the organization. Only then can resilience move from rhetoric to reality, and only then can your PQS become not just a bulwark against failure, but a platform for continuous, serendipitous improvement.
The pharmaceutical industry has long operated under a fundamental epistemological fallacy that undermines our ability to truly understand the effectiveness of our quality systems. We celebrate zero deviations, zero recalls, zero adverse events, and zero regulatory observations as evidence that our systems are working. But a fundamental fact we tend to ignore is that we are confusing the absence of evidence with evidence of absence—a logical error that not only fails to prove effectiveness but actively impedes our ability to build more robust, science-based quality systems.
This challenge strikes at the heart of how we approach quality risk management. When our primary evidence of “success” is that nothing bad happened, we create unfalsifiable systems that can never truly be proven wrong.
The Philosophical Foundation: Falsifiability in Quality Risk Management
Karl Popper’s theory of falsification fundamentally challenges how we think about scientific validity. For Popper, the distinguishing characteristic of genuine scientific theories is not that they can be proven true, but that they can be proven false. A theory that cannot conceivably be refuted by any possible observation is not scientific—it’s metaphysical speculation.
Applied to quality risk management, this creates an uncomfortable truth: most of our current approaches to demonstrating system effectiveness are fundamentally unscientific. When we design quality systems around preventing negative outcomes and then use the absence of those outcomes as evidence of effectiveness, we create what Popper would call unfalsifiable propositions. No possible observation could ever prove our system ineffective as long as we frame effectiveness in terms of what didn’t happen.
Consider the typical pharmaceutical quality narrative: “Our manufacturing process is validated because we haven’t had any quality failures in twelve months.” This statement is unfalsifiable because it can always accommodate new information. If a failure occurs next month, we simply adjust our understanding of the system’s reliability without questioning the fundamental assumption that absence of failure equals validation. We might implement corrective actions, but we rarely question whether our original validation approach was capable of detecting the problems that eventually manifested.
Most of our current risk models are either highly predictive but untestable (making them useful for operational decisions but scientifically questionable) or neither predictive nor testable (making them primarily compliance exercises). The goal should be to move toward models are both scientifically rigorous and practically useful.
This philosophical foundation has practical implications for how we design and evaluate quality risk management systems. Instead of asking “How can we prevent bad things from happening?” we should be asking “How can we design systems that will fail in predictable ways when our underlying assumptions are wrong?” The first question leads to unfalsifiable defensive strategies; the second leads to falsifiable, scientifically valid approaches to quality assurance.
Why “Nothing Bad Happened” Isn’t Evidence of Effectiveness
The fundamental problem with using negative evidence to prove positive claims extends far beyond philosophical niceties, it creates systemic blindness that prevents us from understanding what actually drives quality outcomes. When we frame effectiveness in terms of absence, we lose the ability to distinguish between systems that work for the right reasons and systems that appear to work due to luck, external factors, or measurement limitations.
Scenario
Null Hypothesis
What Rejection Proves
What Non-Rejection Proves
Popperian Assessment
Traditional Efficacy Testing
No difference between treatment and control
Treatment is effective
Cannot prove effectiveness
Falsifiable and useful
Traditional Safety Testing
No increased risk
Treatment increases risk
Cannot prove safety
Unfalsifiable for safety
Absence of Events (Current)
No safety signal detected
Cannot prove anything
Cannot prove safety
Unfalsifiable
Non-inferiority Approach
Excess risk > acceptable margin
Treatment is acceptably safe
Cannot prove safety
Partially falsifiable
Falsification-Based Safety
Safety controls are inadequate
Current safety measures fail
Safety controls are adequate
Falsifiable and actionable
The table above demonstrates how traditional safety and effectiveness assessments fall into unfalsifiable categories. Traditional safety testing, for example, attempts to prove that something doesn’t increase risk, but this can never be definitively demonstrated—we can only fail to detect increased risk within the limitations of our study design. This creates a false confidence that may not be justified by the actual evidence.
The Sampling Illusion: When we observe zero deviations in a batch of 1000 units, we often conclude that our process is in control. But this conclusion conflates statistical power with actual system performance. With typical sampling strategies, we might have only 10% power to detect a 1% defect rate. The “zero observations” reflect our measurement limitations, not process capability.
The Survivorship Bias: Systems that appear effective may be surviving not because they’re well-designed, but because they haven’t yet encountered the conditions that would reveal their weaknesses. Our quality systems are often validated under ideal conditions and then extrapolated to real-world operations where different failure modes may dominate.
The Attribution Problem: When nothing bad happens, we attribute success to our quality systems without considering alternative explanations. Market forces, supplier improvements, regulatory changes, or simple random variation might be the actual drivers of observed outcomes.
Observable Outcome
Traditional Interpretation
Popperian Critique
What We Actually Know
Testable Alternative
Zero adverse events in 1000 patients
“The drug is safe”
Absence of evidence does not equal Evidence of absence
No events detected in this sample
Test limits of safety margin
Zero manufacturing deviations in 12 months
“The process is in control”
No failures observed does not equal a Failure-proof system
No deviations detected with current methods
Challenge process with stress conditions
Zero regulatory observations
“The system is compliant”
No findings does not equal No problems exist
No issues found during inspection
Audit against specific failure modes
Zero product recalls
“Quality is assured”
No recalls does not equal No quality issues
No quality failures reached market
Test recall procedures and detection
Zero patient complaints
“Customer satisfaction achieved”
No complaints does not equal No problems
No complaints received through channels
Actively solicit feedback mechanisms
This table illustrates how traditional interpretations of “positive” outcomes (nothing bad happened) fail to provide actionable knowledge. The Popperian critique reveals that these observations tell us far less than we typically assume, and the testable alternatives provide pathways toward more rigorous evaluation of system effectiveness.
The pharmaceutical industry’s reliance on these unfalsifiable approaches creates several downstream problems. First, it prevents genuine learning and improvement because we can’t distinguish effective interventions from ineffective ones. Second, it encourages defensive mindsets that prioritize risk avoidance over value creation. Third, it undermines our ability to make resource allocation decisions based on actual evidence of what works.
The Model Usefulness Problem: When Predictions Don’t Match Reality
George Box’s famous aphorism that “all models are wrong, but some are useful” provides a pragmatic framework for this challenge, but it doesn’t resolve the deeper question of how to determine when a model has crossed from “useful” to “misleading.” Popper’s falsifiability criterion offers one approach: useful models should make specific, testable predictions that could potentially be proven wrong by future observations.
The challenge in pharmaceutical quality management is that our models often serve multiple purposes that may be in tension with each other. Models used for regulatory submission need to demonstrate conservative estimates of risk to ensure patient safety. Models used for operational decision-making need to provide actionable insights for process optimization. Models used for resource allocation need to enable comparison of risks across different areas of the business.
When the same model serves all these purposes, it often fails to serve any of them well. Regulatory models become so conservative that they provide little guidance for actual operations. Operational models become so complex that they’re difficult to validate or falsify. Resource allocation models become so simplified that they obscure important differences in risk characteristics.
The solution isn’t to abandon modeling, but to be more explicit about the purpose each model serves and the criteria by which its usefulness should be judged. For regulatory purposes, conservative models that err on the side of safety may be appropriate even if they systematically overestimate risks. For operational decision-making, models should be judged primarily on their ability to correctly rank-order interventions by their impact on relevant outcomes. For scientific understanding, models should be designed to make falsifiable predictions that can be tested through controlled experiments or systematic observation.
Consider the example of cleaning validation, where we use models to predict the probability of cross-contamination between manufacturing campaigns. Traditional approaches focus on demonstrating that residual contamination levels are below acceptance criteria—essentially proving a negative. But this approach tells us nothing about the relative importance of different cleaning parameters, the margin of safety in our current procedures, or the conditions under which our cleaning might fail.
A more falsifiable approach would make specific predictions about how changes in cleaning parameters affect contamination levels. We might hypothesize that doubling the rinse time reduces contamination by 50%, or that certain product sequences create systematically higher contamination risks. These hypotheses can be tested and potentially falsified, providing genuine learning about the underlying system behavior.
From Defensive to Testable Risk Management
The evolution from defensive to testable risk management represents a fundamental shift in how we conceptualize quality systems. Traditional defensive approaches ask, “How can we prevent failures?” Testable approaches ask, “How can we design systems that fail predictably when our assumptions are wrong?” This shift moves us from unfalsifiable defensive strategies toward scientifically rigorous quality management.
This transition aligns with the broader evolution in risk thinking documented in ICH Q9(R1) and ISO 31000, which recognize risk as “the effect of uncertainty on objectives” where that effect can be positive, negative, or both. By expanding our definition of risk to include opportunities as well as threats, we create space for falsifiable hypotheses about system performance.
The integration of opportunity-based thinking with Popperian falsifiability creates powerful synergies. When we hypothesize that a particular quality intervention will not only reduce defects but also improve efficiency, we create multiple testable predictions. If the intervention reduces defects but doesn’t improve efficiency, we learn something important about the underlying system mechanics. If it improves efficiency but doesn’t reduce defects, we gain different insights. If it does neither, we discover that our fundamental understanding of the system may be flawed.
This approach requires a cultural shift from celebrating the absence of problems to celebrating the presence of learning. Organizations that embrace falsifiable quality management actively seek conditions that would reveal the limitations of their current systems. They design experiments to test the boundaries of their process capabilities. They view unexpected results not as failures to be explained away, but as opportunities to refine their understanding of system behavior.
The practical implementation of testable risk management involves several key elements:
Hypothesis-Driven Validation: Instead of demonstrating that processes meet specifications, validation activities should test specific hypotheses about process behavior. For example, rather than proving that a sterilization cycle achieves a 6-log reduction, we might test the hypothesis that cycle modifications affect sterility assurance in predictable ways. Instead of demonstrating that the CHO cell culture process consistently produces mAb drug substance meeting predetermined specifications, hypothesis-driven validation would test the specific prediction that maintaining pH at 7.0 ± 0.05 during the production phase will result in final titers that are 15% ± 5% higher than pH maintained at 6.9 ± 0.05, creating a falsifiable hypothesis that can be definitively proven wrong if the predicted titer improvement fails to materialize within the specified confidence intervals
Falsifiable Control Strategies: Control strategies should include specific predictions about how the system will behave under different conditions. These predictions should be testable and potentially falsifiable through routine monitoring or designed experiments.
Learning-Oriented Metrics: Key indicators should be designed to detect when our assumptions about system behavior are incorrect, not just when systems are performing within specification. Metrics that only measure compliance tell us nothing about the underlying system dynamics.
Proactive Stress Testing: Rather than waiting for problems to occur naturally, we should actively probe the boundaries of system performance through controlled stress conditions. This approach reveals failure modes before they impact patients while providing valuable data about system robustness.
Designing Falsifiable Quality Systems
The practical challenge of designing falsifiable quality systems requires a fundamental reconceptualization of how we approach quality assurance. Instead of building systems designed to prevent all possible failures, we need systems designed to fail in instructive ways when our underlying assumptions are incorrect.
This approach starts with making our assumptions explicit and testable. Traditional quality systems often embed numerous unstated assumptions about process behavior, material characteristics, environmental conditions, and human performance. These assumptions are rarely articulated clearly enough to be tested, making the systems inherently unfalsifiable. A falsifiable quality system makes these assumptions explicit and designs tests to evaluate their validity.
Consider the design of a typical pharmaceutical manufacturing process. Traditional approaches focus on demonstrating that the process consistently produces product meeting specifications under defined conditions. This demonstration typically involves process validation studies that show the process works under idealized conditions, followed by ongoing monitoring to detect deviations from expected performance.
A falsifiable approach would start by articulating specific hypotheses about what drives process performance. We might hypothesize that product quality is primarily determined by three critical process parameters, that these parameters interact in predictable ways, and that environmental variations within specified ranges don’t significantly impact these relationships. Each of these hypotheses can be tested and potentially falsified through designed experiments or systematic observation of process performance.
The key insight is that falsifiable quality systems are designed around testable theories of what makes quality systems effective, rather than around defensive strategies for preventing all possible problems. This shift enables genuine learning and continuous improvement because we can distinguish between interventions that work for the right reasons and those that appear to work for unknown or incorrect reasons.
Structured Hypothesis Formation: Quality requirements should be built around explicit hypotheses about cause-and-effect relationships in critical processes. These hypotheses should be specific enough to be tested and potentially falsified through systematic observation or experimentation.
Predictive Monitoring: Instead of monitoring for compliance with specifications, systems should monitor for deviations from predicted behavior. When predictions prove incorrect, this provides valuable information about the accuracy of our underlying process understanding.
Experimental Integration: Routine operations should be designed to provide ongoing tests of system hypotheses. Process changes, material variations, and environmental fluctuations should be treated as natural experiments that provide data about system behavior rather than disturbances to be minimized.
Failure Mode Anticipation: Quality systems should explicitly anticipate the ways failures might happen and design detection mechanisms for these failure modes. This proactive approach contrasts with reactive systems that only detect problems after they occur.
The Evolution of Risk Assessment: From Compliance to Science
The evolution of pharmaceutical risk assessment from compliance-focused activities to genuine scientific inquiry represents one of the most significant opportunities for improving quality outcomes. Traditional risk assessments often function primarily as documentation exercises designed to satisfy regulatory requirements rather than tools for genuine learning and improvement.
ICH Q9(R1) recognizes this limitation and calls for more scientifically rigorous approaches to quality risk management. The updated guidance emphasizes the need for risk assessments to be based on scientific knowledge and to provide actionable insights for quality improvement. This represents a shift away from checklist-based compliance activities toward hypothesis-driven scientific inquiry.
The integration of falsifiability principles with ICH Q9(R1) requirements creates opportunities for more rigorous and useful risk assessments. Instead of asking generic questions about what could go wrong, falsifiable risk assessments develop specific hypotheses about failure modes and design tests to evaluate these hypotheses. This approach provides more actionable insights while meeting regulatory expectations for systematic risk evaluation.
Consider the evolution of Failure Mode and Effects Analysis (FMEA) from a traditional compliance tool to a falsifiable risk assessment method. Traditional FMEA often devolves into generic lists of potential failures with subjective probability and impact assessments. The results provide limited insight because the assessments can’t be systematically tested or validated.
A falsifiable FMEA would start with specific hypotheses about failure mechanisms and their relationships to process parameters, material characteristics, or operational conditions. These hypotheses would be tested through historical data analysis, designed experiments, or systematic monitoring programs. The results would provide genuine insights into system behavior while creating a foundation for continuous improvement.
This evolution requires changes in how we approach several key risk assessment activities:
Hazard Identification: Instead of brainstorming all possible things that could go wrong, risk identification should focus on developing testable hypotheses about specific failure mechanisms and their triggers.
Risk Analysis: Probability and impact assessments should be based on testable models of system behavior rather than subjective expert judgment. When models prove inaccurate, this provides valuable information about the need to revise our understanding of system dynamics.
Risk Control: Control measures should be designed around testable theories of how interventions affect system behavior. The effectiveness of controls should be evaluated through systematic monitoring and periodic testing rather than assumed based on their implementation.
Risk Review: Risk review activities should focus on testing the accuracy of previous risk predictions and updating risk models based on new evidence. This creates a learning loop that continuously improves the quality of risk assessments over time.
Practical Framework for Falsifiable Quality Risk Management
The implementation of falsifiable quality risk management requires a systematic framework that integrates Popperian principles with practical pharmaceutical quality requirements. This framework must be sophisticated enough to generate genuine scientific insights while remaining practical for routine quality management activities.
The foundation of this framework rests on the principle that effective quality systems are built around testable theories of what drives quality outcomes. These theories should make specific predictions that can be evaluated through systematic observation, controlled experimentation, or historical data analysis. When predictions prove incorrect, this provides valuable information about the need to revise our understanding of system behavior.
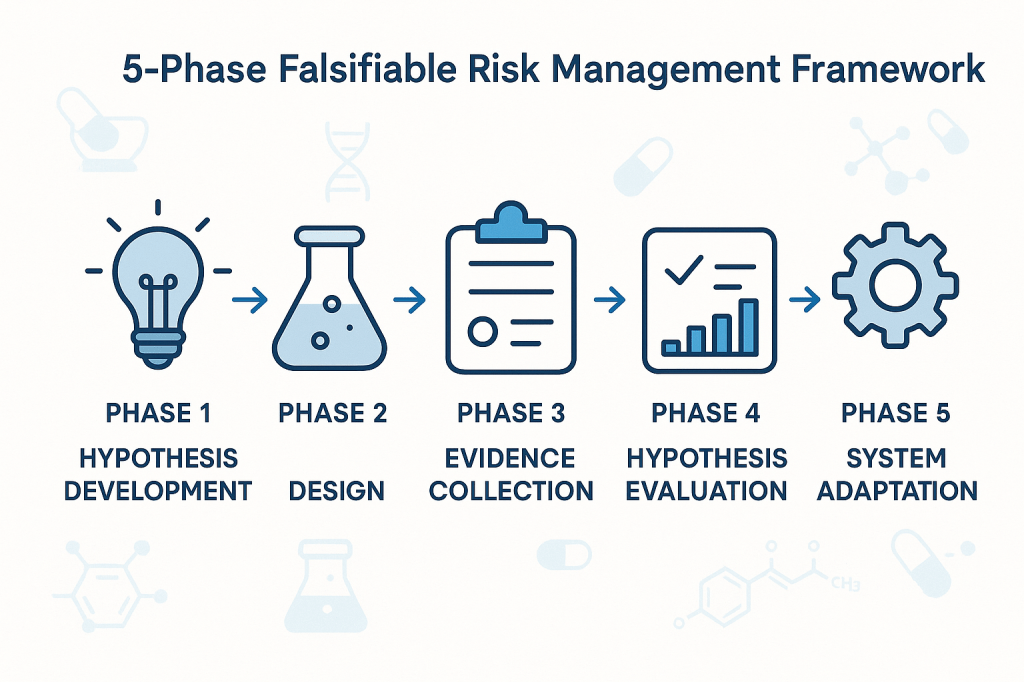
Phase 1: Hypothesis Development
The first phase involves developing specific, testable hypotheses about system behavior. These hypotheses should address fundamental questions about what drives quality outcomes in specific operational contexts. Rather than generic statements about quality risks, hypotheses should make specific predictions about relationships between process parameters, material characteristics, environmental conditions, and quality outcomes.
For example, instead of the generic hypothesis that “temperature variations affect product quality,” a falsifiable hypothesis might state that “temperature excursions above 25°C for more than 30 minutes during the mixing phase increase the probability of out-of-specification results by at least 20%.” This hypothesis is specific enough to be tested and potentially falsified through systematic data collection and analysis.
Phase 2: Experimental Design
The second phase involves designing systematic approaches to test the hypotheses developed in Phase 1. This might involve controlled experiments, systematic analysis of historical data, or structured monitoring programs designed to capture relevant data about hypothesis validity.
The key principle is that testing approaches should be capable of falsifying the hypotheses if they are incorrect. This requires careful attention to statistical power, measurement systems, and potential confounding factors that might obscure true relationships between variables.
Phase 3: Evidence Collection
The third phase focuses on systematic collection of evidence relevant to hypothesis testing. This evidence might come from designed experiments, routine monitoring data, or systematic analysis of historical performance. The critical requirement is that evidence collection should be structured around hypothesis testing rather than generic performance monitoring.
Evidence collection systems should be designed to detect when hypotheses are incorrect, not just when systems are performing within specifications. This requires more sophisticated approaches to data analysis and interpretation than traditional compliance-focused monitoring.
Phase 4: Hypothesis Evaluation
The fourth phase involves systematic evaluation of evidence against the hypotheses developed in Phase 1. This evaluation should follow rigorous statistical methods and should be designed to reach definitive conclusions about hypothesis validity whenever possible.
When hypotheses are falsified, this provides valuable information about the need to revise our understanding of system behavior. When hypotheses are supported by evidence, this provides confidence in our current understanding while suggesting areas for further testing and refinement.
Phase 5: System Adaptation
The final phase involves adapting quality systems based on the insights gained through hypothesis testing. This might involve modifying control strategies, updating risk assessments, or redesigning monitoring programs based on improved understanding of system behavior.
The critical principle is that system adaptations should be based on genuine learning about system behavior rather than reactive responses to compliance issues or external pressures. This creates a foundation for continuous improvement that builds cumulative knowledge about what drives quality outcomes.
Implementation Challenges
The transition to falsifiable quality risk management faces several practical challenges that must be addressed for successful implementation. These challenges range from technical issues related to experimental design and statistical analysis to cultural and organizational barriers that may resist more scientifically rigorous approaches to quality management.
Technical Challenges
The most immediate technical challenge involves designing falsifiable hypotheses that are relevant to pharmaceutical quality management. Many quality professionals have extensive experience with compliance-focused activities but limited experience with experimental design and hypothesis testing. This skills gap must be addressed through targeted training and development programs.
Statistical power represents another significant technical challenge. Many quality systems operate with very low baseline failure rates, making it difficult to design experiments with adequate power to detect meaningful differences in system performance. This requires sophisticated approaches to experimental design and may necessitate longer observation periods or larger sample sizes than traditionally used in quality management.
Measurement systems present additional challenges. Many pharmaceutical quality attributes are difficult to measure precisely, introducing uncertainty that can obscure true relationships between process parameters and quality outcomes. This requires careful attention to measurement system validation and uncertainty quantification.
Cultural and Organizational Challenges
Perhaps more challenging than technical issues are the cultural and organizational barriers to implementing more scientifically rigorous quality management approaches. Many pharmaceutical organizations have deeply embedded cultures that prioritize risk avoidance and compliance over learning and improvement.
The shift to falsifiable quality management requires cultural change that embraces controlled failure as a learning opportunity rather than something to be avoided at all costs. This represents a fundamental change in how many organizations think about quality management and may encounter significant resistance.
Regulatory relationships present additional organizational challenges. Many quality professionals worry that more rigorous scientific approaches to quality management might raise regulatory concerns or create compliance burdens. This requires careful communication with regulatory agencies to demonstrate that falsifiable approaches enhance rather than compromise patient safety.
Strategic Solutions
Successfully implementing falsifiable quality risk management requires strategic approaches that address both technical and cultural challenges. These solutions must be tailored to specific organizational contexts while maintaining scientific rigor and regulatory compliance.
Pilot Programs: Implementation should begin with carefully selected pilot programs in areas where falsifiable approaches can demonstrate clear value. These pilots should be designed to generate success stories that support broader organizational adoption while building internal capability and confidence.
Training and Development: Comprehensive training programs should be developed to build organizational capability in experimental design, statistical analysis, and hypothesis testing. These programs should be tailored to pharmaceutical quality contexts and should emphasize practical applications rather than theoretical concepts.
Regulatory Engagement: Proactive engagement with regulatory agencies should emphasize how falsifiable approaches enhance patient safety through improved understanding of system behavior. This communication should focus on the scientific rigor of the approach rather than on business benefits that might appear secondary to regulatory objectives.
Cultural Change Management: Systematic change management programs should address cultural barriers to embracing controlled failure as a learning opportunity. These programs should emphasize how falsifiable approaches support regulatory compliance and patient safety rather than replacing these priorities with business objectives.
Case Studies: Falsifiability in Practice
The practical application of falsifiable quality risk management can be illustrated through several case studies that demonstrate how Popperian principles can be integrated with routine pharmaceutical quality activities. These examples show how hypotheses can be developed, tested, and used to improve quality outcomes while maintaining regulatory compliance.
Case Study 1: Cleaning Validation Optimization
A biologics manufacturer was experiencing occasional cross-contamination events despite having validated cleaning procedures that consistently met acceptance criteria. Traditional approaches focused on demonstrating that cleaning procedures reduced contamination below specified limits, but provided no insight into the factors that occasionally caused this system to fail.
The falsifiable approach began with developing specific hypotheses about cleaning effectiveness. The team hypothesized that cleaning effectiveness was primarily determined by three factors: contact time with cleaning solution, mechanical action intensity, and rinse water temperature. They further hypothesized that these factors interacted in predictable ways and that current procedures provided a specific margin of safety above minimum requirements.
These hypotheses were tested through a designed experiment that systematically varied each cleaning parameter while measuring residual contamination levels. The results revealed that current procedures were adequate under ideal conditions but provided minimal margin of safety when multiple factors were simultaneously at their worst-case levels within specified ranges.
Based on these findings, the cleaning procedure was modified to provide greater margin of safety during worst-case conditions. More importantly, ongoing monitoring was redesigned to test the continued validity of the hypotheses about cleaning effectiveness rather than simply verifying compliance with acceptance criteria.
Case Study 2: Process Control Strategy Development
A pharmaceutical manufacturer was developing a control strategy for a new manufacturing process. Traditional approaches would have focused on identifying critical process parameters and establishing control limits based on process validation studies. Instead, the team used a falsifiable approach that started with explicit hypotheses about process behavior.
The team hypothesized that product quality was primarily controlled by the interaction between temperature and pH during the reaction phase, that these parameters had linear effects on product quality within the normal operating range, and that environmental factors had negligible impact on these relationships.
These hypotheses were tested through systematic experimentation during process development. The results confirmed the importance of the temperature-pH interaction but revealed nonlinear effects that weren’t captured in the original hypotheses. More importantly, environmental humidity was found to have significant effects on process behavior under certain conditions.
The control strategy was designed around the revised understanding of process behavior gained through hypothesis testing. Ongoing process monitoring was structured to continue testing key assumptions about process behavior rather than simply detecting deviations from target conditions.
Case Study 3: Supplier Quality Management
A biotechnology company was managing quality risks from a critical raw material supplier. Traditional approaches focused on incoming inspection and supplier auditing to verify compliance with specifications and quality system requirements. However, occasional quality issues suggested that these approaches weren’t capturing all relevant quality risks.
The falsifiable approach started with specific hypotheses about what drove supplier quality performance. The team hypothesized that supplier quality was primarily determined by their process control during critical manufacturing steps, that certain environmental conditions increased the probability of quality issues, and that supplier quality system maturity was predictive of long-term quality performance.
These hypotheses were tested through systematic analysis of supplier quality data, enhanced supplier auditing focused on specific process control elements, and structured data collection about environmental conditions during material manufacturing. The results revealed that traditional quality system assessments were poor predictors of actual quality performance, but that specific process control practices were strongly predictive of quality outcomes.
The supplier management program was redesigned around the insights gained through hypothesis testing. Instead of generic quality system requirements, the program focused on specific process control elements that were demonstrated to drive quality outcomes. Supplier performance monitoring was structured around testing continued validity of the relationships between process control and quality outcomes.
Measuring Success in Falsifiable Quality Systems
The evaluation of falsifiable quality systems requires fundamentally different approaches to performance measurement than traditional compliance-focused systems. Instead of measuring the absence of problems, we need to measure the presence of learning and the accuracy of our predictions about system behavior.
Traditional quality metrics focus on outcomes: defect rates, deviation frequencies, audit findings, and regulatory observations. While these metrics remain important for regulatory compliance and business performance, they provide limited insight into whether our quality systems are actually effective or merely lucky. Falsifiable quality systems require additional metrics that evaluate the scientific validity of our approach to quality management.
Predictive Accuracy Metrics
The most direct measure of a falsifiable quality system’s effectiveness is the accuracy of its predictions about system behavior. These metrics evaluate how well our hypotheses about quality system behavior match observed outcomes. High predictive accuracy suggests that we understand the underlying drivers of quality outcomes. Low predictive accuracy indicates that our understanding needs refinement.
Predictive accuracy metrics might include the percentage of process control predictions that prove correct, the accuracy of risk assessments in predicting actual quality issues, or the correlation between predicted and observed responses to process changes. These metrics provide direct feedback about the validity of our theoretical understanding of quality systems.
Learning Rate Metrics
Another important category of metrics evaluates how quickly our understanding of quality systems improves over time. These metrics measure the rate at which falsified hypotheses lead to improved system performance or more accurate predictions. High learning rates indicate that the organization is effectively using falsifiable approaches to improve quality outcomes.
Learning rate metrics might include the time required to identify and correct false assumptions about system behavior, the frequency of successful process improvements based on hypothesis testing, or the rate of improvement in predictive accuracy over time. These metrics evaluate the dynamic effectiveness of falsifiable quality management approaches.
Hypothesis Quality Metrics
The quality of hypotheses generated by quality risk management processes represents another important performance dimension. High-quality hypotheses are specific, testable, and relevant to important quality outcomes. Poor-quality hypotheses are vague, untestable, or focused on trivial aspects of system performance.
Hypothesis quality can be evaluated through structured peer review processes, assessment of testability and specificity, and evaluation of relevance to critical quality attributes. Organizations with high-quality hypothesis generation processes are more likely to gain meaningful insights from their quality risk management activities.
System Robustness Metrics
Falsifiable quality systems should become more robust over time as learning accumulates and system understanding improves. Robustness can be measured through the system’s ability to maintain performance despite variations in operating conditions, changes in materials or equipment, or other sources of uncertainty.
Robustness metrics might include the stability of process performance across different operating conditions, the effectiveness of control strategies under stress conditions, or the system’s ability to detect and respond to emerging quality risks. These metrics evaluate whether falsifiable approaches actually lead to more reliable quality systems.
Regulatory Implications and Opportunities
The integration of falsifiable principles with pharmaceutical quality risk management creates both challenges and opportunities in regulatory relationships. While some regulatory agencies may initially view scientific approaches to quality management with skepticism, the ultimate result should be enhanced regulatory confidence in quality systems that can demonstrate genuine understanding of what drives quality outcomes.
The key to successful regulatory engagement lies in emphasizing how falsifiable approaches enhance patient safety rather than replacing regulatory compliance with business optimization. Regulatory agencies are primarily concerned with patient safety and product quality. Falsifiable quality systems support these objectives by providing more rigorous and reliable approaches to ensuring quality outcomes.
Enhanced Regulatory Submissions
Regulatory submissions based on falsifiable quality systems can provide more compelling evidence of system effectiveness than traditional compliance-focused approaches. Instead of demonstrating that systems meet minimum requirements, falsifiable approaches can show genuine understanding of what drives quality outcomes and how systems will behave under different conditions.
This enhanced evidence can support regulatory flexibility in areas such as process validation, change control, and ongoing monitoring requirements. Regulatory agencies may be willing to accept risk-based approaches to these activities when they’re supported by rigorous scientific evidence rather than generic compliance activities.
Proactive Risk Communication
Falsifiable quality systems enable more proactive and meaningful communication with regulatory agencies about quality risks and mitigation strategies. Instead of reactive communication about compliance issues, organizations can engage in scientific discussions about system behavior and improvement strategies.
This proactive communication can build regulatory confidence in organizational quality management capabilities while providing opportunities for regulatory agencies to provide input on scientific approaches to quality improvement. The result should be more collaborative regulatory relationships based on shared commitment to scientific rigor and patient safety.
Regulatory Science Advancement
The pharmaceutical industry’s adoption of more scientifically rigorous approaches to quality management can contribute to the advancement of regulatory science more broadly. Regulatory agencies benefit from industry innovations in risk assessment, process understanding, and quality assurance methods.
Organizations that successfully implement falsifiable quality risk management can serve as case studies for regulatory guidance development and can provide evidence for the effectiveness of science-based approaches to quality assurance. This contribution to regulatory science advancement creates value that extends beyond individual organizational benefits.
Toward a More Scientific Quality Culture
The long-term vision for falsifiable quality risk management extends beyond individual organizational implementations to encompass fundamental changes in how the pharmaceutical industry approaches quality assurance. This vision includes more rigorous scientific approaches to quality management, enhanced collaboration between industry and regulatory agencies, and continuous advancement in our understanding of what drives quality outcomes.
Industry-Wide Learning Networks
One promising direction involves the development of industry-wide learning networks that share insights from falsifiable quality management implementations. These networks facilitate collaborative hypothesis testing, shared learning from experimental results, and development of common methodologies for scientific approaches to quality assurance.
Such networks accelerate the advancement of quality science while maintaining appropriate competitive boundaries. Organizations should share methodological insights and general findings while protecting proprietary information about specific processes or products. The result would be faster advancement in quality management science that benefits the entire industry.
Advanced Analytics Integration
The integration of advanced analytics and machine learning techniques with falsifiable quality management approaches represents another promising direction. These technologies can enhance our ability to develop testable hypotheses, design efficient experiments, and analyze complex datasets to evaluate hypothesis validity.
Machine learning approaches are particularly valuable for identifying patterns in complex quality datasets that might not be apparent through traditional analysis methods. However, these approaches must be integrated with falsifiable frameworks to ensure that insights can be validated and that predictive models can be systematically tested and improved.
Regulatory Harmonization
The global harmonization of regulatory approaches to science-based quality management represents a significant opportunity for advancing patient safety and regulatory efficiency. As individual regulatory agencies gain experience with falsifiable quality management approaches, there are opportunities to develop harmonized guidance that supports consistent global implementation.
ICH Q9(r1) was a great step. I would love to see continued work in this area.
Embracing the Discomfort of Scientific Rigor
The transition from compliance-focused to scientifically rigorous quality risk management represents more than a methodological change—it requires fundamentally rethinking how we approach quality assurance in pharmaceutical manufacturing. By embracing Popper’s challenge that genuine scientific theories must be falsifiable, we move beyond the comfortable but ultimately unhelpful world of proving negatives toward the more demanding but ultimately more rewarding world of testing positive claims about system behavior.
The effectiveness paradox that motivates this discussion—the problem of determining what works when our primary evidence is that “nothing bad happened”—cannot be resolved through better compliance strategies or more sophisticated documentation. It requires genuine scientific inquiry into the mechanisms that drive quality outcomes. This inquiry must be built around testable hypotheses that can be proven wrong, not around defensive strategies that can always accommodate any possible outcome.
The practical implementation of falsifiable quality risk management is not without challenges. It requires new skills, different cultural approaches, and more sophisticated methodologies than traditional compliance-focused activities. However, the potential benefits—genuine learning about system behavior, more reliable quality outcomes, and enhanced regulatory confidence—justify the investment required for successful implementation.
Perhaps most importantly, the shift to falsifiable quality management moves us toward a more honest assessment of what we actually know about quality systems versus what we merely assume or hope to be true. This honesty is uncomfortable but essential for building quality systems that genuinely serve patient safety rather than organizational comfort.
The question is not whether pharmaceutical quality management will eventually embrace more scientific approaches—the pressures of regulatory evolution, competitive dynamics, and patient safety demands make this inevitable. The question is whether individual organizations will lead this transition or be forced to follow. Those that embrace the discomfort of scientific rigor now will be better positioned to thrive in a future where quality management is evaluated based on genuine effectiveness rather than compliance theater.
As we continue to navigate an increasingly complex regulatory and competitive environment, the organizations that master the art of turning uncertainty into testable knowledge will be best positioned to deliver consistent quality outcomes while maintaining the flexibility needed for innovation and continuous improvement. The integration of Popperian falsifiability with modern quality risk management provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.
The path forward requires courage to question our current assumptions, discipline to design rigorous tests of our theories, and wisdom to learn from both our successes and our failures. But for those willing to embrace these challenges, the reward is quality systems that are not only compliant but genuinely effective. Systems that we can defend not because they’ve never been proven wrong, but because they’ve been proven right through systematic, scientific inquiry.