Ineffective risk management and quality systems revolve around superficial risk management. The core issue? Teams designed for compliance as a check-the-box activity rather than cognitive rigor. These gaps create systematic blind spots that no checklist can fix. The solution isn’t more assessors—it’s fewer, more competent ones anchored in science, patient impact, and lived process reality.
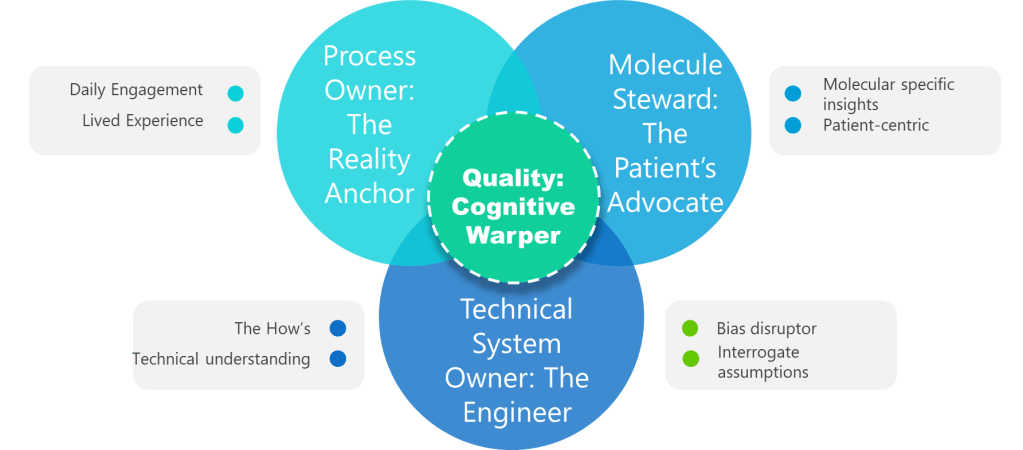
Not a title. A lived experience. Superficial ownership creates the “unjustified assumptions.” This role requires daily engagement with the process—not just signature authority. Without it, assumptions go unchallenged.
Beyond “SME”—the protein whisperer. This role demands provable knowledge of degradation pathways, critical quality attributes (CQAs), and patient impact. Contrast this with generic “subject matter experts” who lack molecule-specific insights. Without this anchor, assessments overlook patient-centric failure modes.
3. Technical System Owner: The Engineer
The value of the Technical System Owner—often the engineer—lies in their unique ability to bridge the worlds of design, operations, and risk control throughout the pharmaceutical lifecycle. Far from being a mere custodian of equipment, the system owner is the architect who understands not just how a system is built, but how it behaves under real-world conditions and how it integrates with the broader manufacturing program
4. Quality: The Cognitive Warper
Forget the auditor—this is your bias disruptor. Quality’s value lies in forcing cross-functional dialogue, challenging tacit assumptions, and documenting debates. When Quality fails to interrogate assumptions, hazards go unidentified. Their real role: Mandate “assumption logs” where every “We’ve always done it this way” must produce data or die.
Team Design as Knowledge Preservation
Team design in the context of risk management is fundamentally an act of knowledge preservation, not just an exercise in filling seats or meeting compliance checklists. Every effective risk team is a living repository of the organization’s critical process insights, technical know-how, and nuanced operational experience. When teams are thoughtfully constructed to include individuals with deep, hands-on familiarity—process owners, technical system engineers, molecule stewards, and quality integrators—they collectively safeguard the hard-won lessons and tacit knowledge that are so often lost when people move on or retire. This approach ensures that risk assessments are not just theoretical exercises but are grounded in the practical realities that only those with lived experience can provide.
Combating organizational forgetting requires more than documentation or digital knowledge bases; it demands intentional, cross-functional team design that fosters active knowledge transfer. When a risk team brings together diverse experts who routinely interact, challenge each other’s assumptions, and share context from their respective domains, they create a dynamic environment where critical information is surfaced, scrutinized, and retained. This living dialogue is far more effective than static records, as it allows for the continuous updating and contextualization of knowledge in response to new challenges, regulatory changes, and operational shifts. In this way, team design becomes a strategic defense against the silent erosion of expertise that can leave organizations exposed to avoidable risks.
Ultimately, investing in team design as a knowledge preservation strategy is about building organizational resilience. It means recognizing that the greatest threats often arise not from what is known, but from what is forgotten or never shared. By prioritizing teams that embody both breadth and depth of experience, organizations create a robust safety net—one that catches subtle warning signs, adapts to evolving risks, and ensures that critical knowledge endures beyond any single individual’s tenure. This is how organizations move from reactive problem-solving to proactive risk management, turning collective memory into a competitive advantage and a foundation for sustained quality.
Call to Action: Build the Risk Team
Moving from compliance theater to true protection starts with assembling a team designed for cognitive rigor, knowledge depth and psychological safety.
Start with a Clear Charter, Not a Checklist
An excellent risk team exists to frame, analyse and communicate uncertainty so that the business can make science-based, patient-centred decisions. Assigning authorities and accountabilities is a leadership duty, not an after-thought. Before naming people, write down:
the decisions the team must enable,
the degree of formality those decisions demand, and
the resources (time, data, tools) management will guarantee.
Without this charter, even star performers will default to box-ticking.
Fill Four Core Seats – And Prove Competence
ICH Q9 is blunt: risk work should be done by interdisciplinary teams that include experts from quality, engineering, operations and regulatory affairs. ASTM E2500 translates that into a requirement for documented subject-matter experts (SMEs) who own critical knowledge throughout the lifecycle. Map those expectations onto four non-negotiable roles.
Process Owner – The Reality Anchor: This individual has lived the operation in the last 90 days, not just signed SOPs. They carry the authority to change methods, budgets and training, and enough hands-on credibility to spot when a theoretical control will never work on the line. Authentic owners dismantle assumptions by grounding every risk statement in current shop-floor facts.
Molecule Steward – The Patient’s Advocate: Too often “SME” is shorthand for “the person available.” The molecule steward is different: a scientist who understands how the specific product fails and can translate deviations into patient impact. When temperature drifts two degrees during freeze-drying, the steward can explain whether a monoclonal antibody will aggregate or merely lose a day of shelf life. Without this anchor, the team inevitably under-scores hazards that never appear in a generic FMEA template.
Technical System Owner – The Engineering Interpreter: Equipment does not care about meeting minutes; it obeys physics. The system owner must articulate functional requirements, design limits and integration logic. Where a tool-focused team may obsess over gasket leaks, the system owner points out that a single-loop PLC has no redundancy and that a brief voltage dip could push an entire batch outside critical parameters—a classic case of method over physics.
Quality Integrator – The Bias Disruptor: Quality’s mission is to force cross-functional dialogue and preserve evidence. That means writing assumption logs, challenging confirmation bias and ensuring that dissenting voices are heard. The quality lead also maintains the knowledge repository so future teams are not condemned to repeat forgotten errors.
Secure Knowledge Accessibility, Not Just Possession
A credentialed expert who cannot be reached when the line is down at 2 a.m. is as useful as no expert at all. Conduct aKnowledge Accessibility Index audit before every major assessment.
Embed Psychological Safety to Unlock the Team’s Brainpower
No amount of SOPs compensates for a culture that punishes bad news. Staff speak up only when leaders are approachable, intolerant of blame and transparent about their own fallibility. Leaders must therefore:
Invite dissent early: begin meetings with “What might we be overlooking?”
Model vulnerability: share personal errors and how the system, not individuals, failed.
Reward candor: recognize the engineer who halted production over a questionable trend.
Choose Methods Last, After Understanding the Science
Excellent teams let the problem dictate the tool, not vice versa. They build a failure-tree or block diagram first, then decide whether FMEA, FTA or bow-tie analysis will illuminate the weak spot. If the team defaults to a method because “it’s in the SOP,” stop and reassess. Tool selection is a decision, not a reflex.
Provide Time and Resources Proportionate to Uncertainty
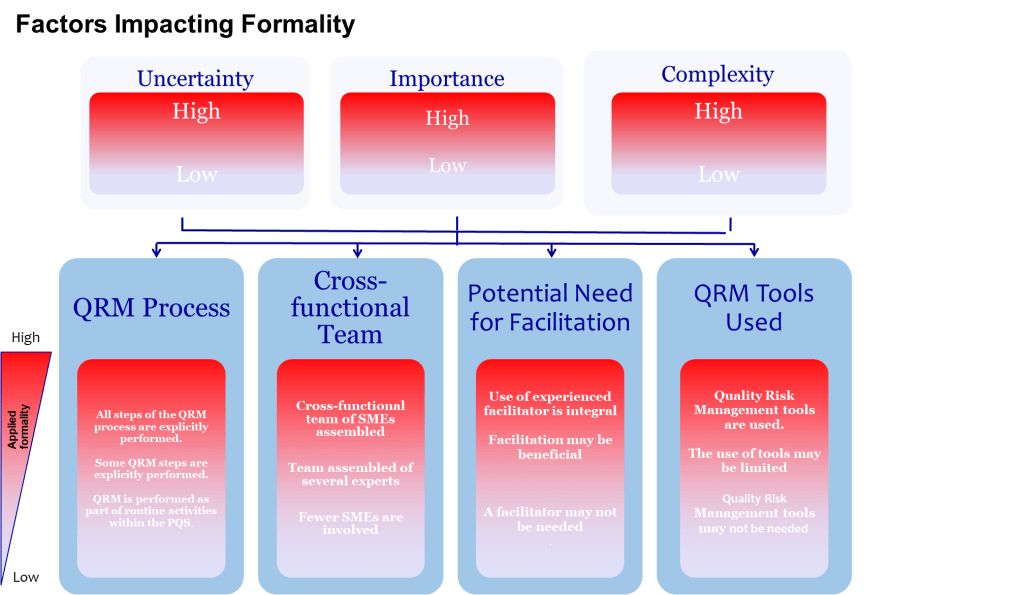
ICH Q9 asks decision-makers to ensure resources match the risk question. Complex, high-uncertainty topics demand longer workshops, more data and external review, while routine changes may only need a rapid check. Resist the urge to shoehorn every assessment into a one-hour meeting because calendars are overloaded.
Institutionalize Learning Loops
Great teams treat every assessment as both analysis and experiment. They:
Track prediction accuracy: did the “medium”-ranked hazard occur?
Compare expected versus actual detectability: were controls as effective as assumed?
Feed insights into updated templates and training so the next team starts smarter.
The loop closes when the knowledge base evolves at the same pace as the plant.
When to Escalate – The Abort-Mission Rule
If a risk scenario involves patient safety, novel technology and the molecule steward is unavailable, stop. The assessment waits until a proper team is in the room. Rushing ahead satisfies schedules, not safety.
Conclusion
Excellence in risk management is rarely about adding headcount; it is about curating brains with complementary lenses and giving them the culture, structure and time to think. Build that environment and the monsters stay on the storyboard, never in the plant.
The Hidden Architecture of Risk Assessment Failure
Peter Baker‘s blunt assessment, “We allowed all these players into the market who never should have been there in the first place, ” hits at something we all recognize but rarely talk about openly. Here’s the uncomfortable truth: even seasoned quality professionals with decades of experience and proven methodologies can miss critical risks that seem obvious in hindsight. Recognizing this truth is not about competence or dedication. It is about acknowledging that our expertise, no matter how extensive, operates within cognitive frameworks that can create blind spots. The real opportunity lies in understanding how these mental patterns shape our decisions and building knowledge systems that help us see what we might otherwise miss. When we’re honest about these limitations, we can strengthen our approaches and create more robust quality systems.
The framework of risk management, designed to help avoid the monsters of bad decision-making, can all too often fail us. Luckily, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidance document PI 038-2 “Assessment of Quality Risk Management Implementation” identifies three critical observations that reveal systematic vulnerabilities in risk management practice: unjustified assumptions, incomplete identification of risks or inadequate information, and lack of relevant experience with inappropriate use of risk assessment tools. These observations represent something more profound than procedural failures—they expose cognitive and knowledge management vulnerabilities that can undermine even the most well-intentioned quality systems..
Understanding these vulnerabilities through the lens of cognitive behavioral science and knowledge management principles provides a pathway to more robust and resilient quality systems. Instead of viewing these failures as isolated incidents or individual shortcomings, we should recognize them as predictable patterns that emerge from systematic limitations in how humans process information and organizations manage knowledge. This recognition opens the door to designing quality systems that work with, rather than against, these cognitive realities
The Framework Foundation of Risk Management Excellence
Risk management operates fundamentally as a frameworkrather than a rigid methodology, providing the structural architecture that enables systematic approaches to identifying, assessing, and controlling uncertainties that could impact pharmaceutical quality objectives. This distinction proves crucial for understanding how cognitive biases manifest within risk management systems and how excellence-driven quality systems can effectively address them.
A framework establishes the high-level structure, principles, and processes for managing risks systematically while allowing flexibility in execution and adaptation to specific organizational contexts. The framework defines structural components like governance and culture, strategy and objective-setting, and performance monitoring that establish the scaffolding for risk management without prescribing inflexible procedures.
Within this framework structure, organizations deploy specific methodological elements as tools for executing particular risk management tasks. These methodologies include techniques such as Failure Mode and Effects Analysis (FMEA), brainstorming sessions, SWOT analysis, and risk surveys for identification activities, while assessment methodologies encompass qualitative and quantitative approaches including statistical models and scenario analysis. The critical insight is that frameworks provide the systematic architecture that counters cognitive biases, while methodologies are specific techniques deployed within this structure.
This framework approach directly addresses the three PIC/S observations by establishing systematic requirements that counter natural cognitive tendencies. Standardized framework processes force systematic consideration of risk factors rather than allowing teams to rely on intuitive pattern recognition that might be influenced by availability bias or anchoring on familiar scenarios. Documented decision rationales required by framework approaches make assumptions explicit and subject to challenge, preventing the perpetuation of unjustified beliefs that may have become embedded in organizational practices.
The governance components inherent in risk management frameworks address the expertise and knowledge management challenges identified in PIC/S guidance by establishing clear roles, responsibilities, and requirements for appropriate expertise involvement in risk assessment activities. Rather than leaving expertise requirements to chance or individual judgment, frameworks systematically define when specialized knowledge is required and how it should be accessed and validated.
ICH Q9’s approach to Quality Risk Management in pharmaceuticals demonstrates this framework principle through its emphasis on scientific knowledge and proportionate formality. The guideline establishes framework requirements that risk assessments be “based on scientific knowledge and linked to patient protection” while allowing methodological flexibility in how these requirements are met. This framework approach provides systematic protection against the cognitive biases that lead to unjustified assumptions while supporting the knowledge management processes necessary for complete risk identification and appropriate tool application.
The continuous improvement cycles embedded in mature risk management frameworks provide ongoing validation of cognitive bias mitigation effectiveness through operational performance data. These systematic feedback loops enable organizations to identify when initial assumptions prove incorrect or when changing conditions alter risk profiles, supporting the adaptive learning required for sustained excellence in pharmaceutical risk management.
The Systematic Nature of Risk Assessment Failure
Unjustified Assumptions: When Experience Becomes Liability
The first PIC/S observation—unjustified assumptions—represents perhaps the most insidious failure mode in pharmaceutical risk management. These are decisions made without sufficient scientific evidence or rational basis, often arising from what appears to be strength: extensive experience with familiar processes. The irony is that the very expertise we rely upon can become a source of systematic error when it leads to unfounded confidence in our understanding.
This phenomenon manifests most clearly in what cognitive scientists call anchoring bias—the tendency to rely too heavily on the first piece of information encountered when making decisions. In pharmaceutical risk assessments, this might appear as teams anchoring on historical performance data without adequately considering how process changes, equipment aging, or supply chain modifications might alter risk profiles. The assumption becomes: “This process has worked safely for five years, so the risk profile remains unchanged.”
Confirmation bias compounds this issue by causing assessors to seek information that confirms their existing beliefs while ignoring contradictory evidence. Teams may unconsciously filter available data to support predetermined conclusions about process reliability or control effectiveness. This creates a self-reinforcing cycle where assumptions become accepted facts, protected from challenge by selective attention to supporting evidence.
The knowledge management dimension of this failure is equally significant. Organizations often lack systematic approaches to capturing and validating the assumptions embedded in institutional knowledge. Tacit knowledge—the experiential, intuitive understanding that experts develop over time—becomes problematic when it remains unexamined and unchallenged. Without explicit processes to surface and test these assumptions, they become invisible constraints on risk assessment effectiveness.
Incomplete Risk Identification: The Boundaries of Awareness
The second observation—incomplete identification of risks or inadequate information—reflects systematic failures in the scope and depth of risk assessment activities. This represents more than simple oversight; it demonstrates how cognitive limitations and organizational boundaries constrain our ability to identify potential hazards comprehensively.
Availability bias plays a central role in this failure mode. Risk assessment teams naturally focus on hazards that are easily recalled or recently experienced, leading to overemphasis on dramatic but unlikely events while underestimating more probable but less memorable risks. A team might spend considerable time analyzing the risk of catastrophic equipment failure while overlooking the cumulative impact of gradual process drift or material variability.
The knowledge management implications are profound. Organizations often struggle with knowledge that exists in isolated pockets of expertise. Critical information about process behaviors, failure modes, or control limitations may be trapped within specific functional areas or individual experts. Without systematic mechanisms to aggregate and synthesize distributed knowledge, risk assessments operate on fundamentally incomplete information.
Groupthink and organizational boundaries further constrain risk identification. When risk assessment teams are composed of individuals from similar backgrounds or organizational levels, they may share common blind spots that prevent recognition of certain hazard categories. The pressure to reach consensus can suppress dissenting views that might identify overlooked risks.
Inappropriate Tool Application: When Methodology Becomes Mythology
The third observation—lack of relevant experience with process assessment and inappropriate use of risk assessment tools—reveals how methodological sophistication can mask fundamental misunderstanding. This failure mode is particularly dangerous because it generates false confidence in risk assessment conclusions while obscuring the limitations of the analysis.
Overconfidence bias drives teams to believe they have more expertise than they actually possess, leading to misapplication of complex risk assessment methodologies. A team might apply Failure Mode and Effects Analysis (FMEA) to a novel process without adequate understanding of either the methodology’s limitations or the process’s unique characteristics. The resulting analysis appears scientifically rigorous while providing misleading conclusions about risk levels and control effectiveness.
This connects directly to knowledge management failures in expertise distribution and access. Organizations may lack systematic approaches to identifying when specialized knowledge is required for risk assessments and ensuring that appropriate expertise is available when needed. The result is risk assessments conducted by well-intentioned teams who lack the specific knowledge required for accurate analysis.
The problem is compounded when organizations rely heavily on external consultants or standardized methodologies without developing internal capabilities for critical evaluation. While external expertise can be valuable, sole reliance on these resources may result in inappropriate conclusions or a lack of ownership of the assessment, as the PIC/S guidance explicitly warns.
The Role of Negative Reasoning in Risk Assessment
The research on causal reasoning versus negative reasoning from Energy Safety Canada provides additional insight into systematic failures in pharmaceutical risk assessments. Traditional root cause analysis often focuses on what did not happen rather than what actually occurred—identifying “counterfactuals” such as “operators not following procedures” or “personnel not stopping work when they should have.”
This approach, termed “negative reasoning,” is fundamentally flawed because what was not happening cannot create the outcomes we experienced. These counterfactuals “exist only in retrospection and never actually influenced events,” yet they dominate many investigation conclusions. In risk assessment contexts, this manifests as teams focusing on the absence of desired behaviors or controls rather than understanding the positive factors that actually influence system performance.
The shift toward causal reasoning requires understanding what actually occurred and what factors positively influenced the outcomes observed.
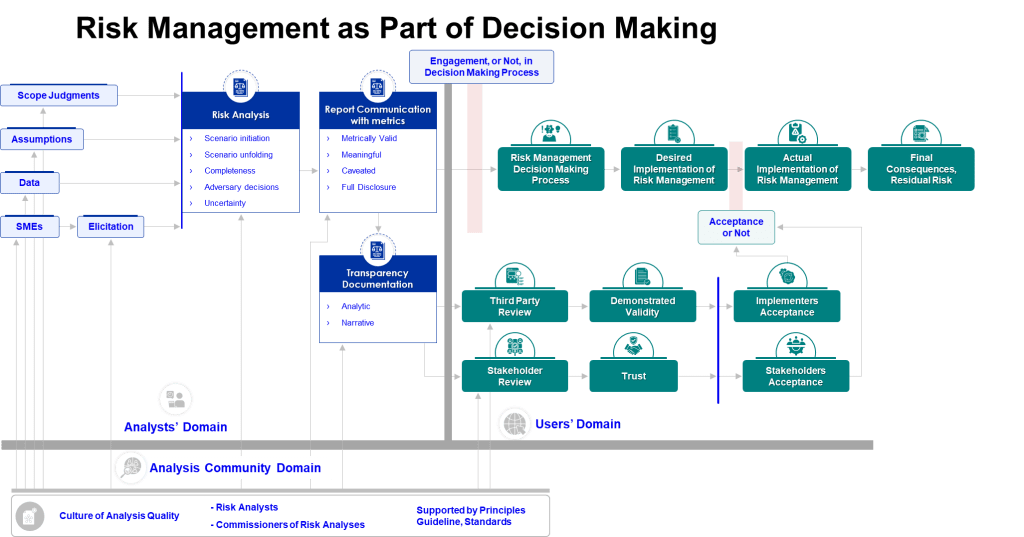
Knowledge-Enabled Decision Making
The intersection of cognitive science and knowledge management reveals how organizations can design systems that support better risk assessment decisions. Knowledge-enabled decision making requires structures that make relevant information accessible at the point of decision while supporting the cognitive processes necessary for accurate analysis.
This involves several key elements:
Structured knowledge capture that explicitly identifies assumptions, limitations, and context for recorded information. Rather than simply documenting conclusions, organizations must capture the reasoning process and evidence base that supports risk assessment decisions.
Knowledge validation systems that systematically test assumptions embedded in organizational knowledge. This includes processes for challenging accepted wisdom and updating mental models when new evidence emerges.
Expertise networks that connect decision-makers with relevant specialized knowledge when required. Rather than relying on generalist teams for all risk assessments, organizations need systematic approaches to accessing specialized expertise when process complexity or novelty demands it.
Decision support systems that prompt systematic consideration of potential biases and alternative explanations.
Excellence and Elegance: Designing Quality Systems for Cognitive Reality
Structured Decision-Making Processes
Excellence in pharmaceutical quality systems requires moving beyond hoping that individuals will overcome cognitive limitations through awareness alone. Instead, organizations must design structured decision-making processes that systematically counter known biases while supporting comprehensive risk identification and analysis.
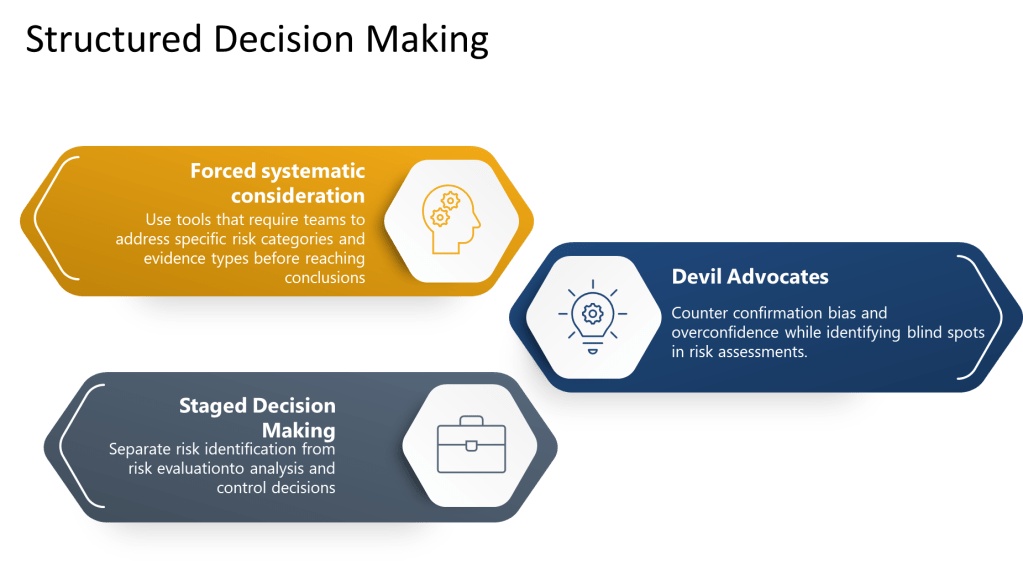
Forced systematic consideration involves using checklists, templates, and protocols that require teams to address specific risk categories and evidence types before reaching conclusions. Rather than relying on free-form discussion that may be influenced by availability bias or groupthink, these tools ensure comprehensive coverage of relevant factors.
Devil’s advocate processes systematically introduce alternative perspectives and challenge preferred conclusions. By assigning specific individuals to argue against prevailing views or identify overlooked risks, organizations can counter confirmation bias and overconfidence while identifying blind spots in risk assessments.
Staged decision-making separates risk identification from risk evaluation, preventing premature closure and ensuring adequate time for comprehensive hazard identification before moving to analysis and control decisions.
Multi-Perspective Analysis and Diverse Assessment Teams
Cognitive diversity in risk assessment teams provides natural protection against individual and group biases. This goes beyond simple functional representation to include differences in experience, training, organizational level, and thinking styles that can identify risks and solutions that homogeneous teams might miss.
Cross-functional integration ensures that risk assessments benefit from different perspectives on process performance, control effectiveness, and potential failure modes. Manufacturing, quality assurance, regulatory affairs, and technical development professionals each bring different knowledge bases and mental models that can reveal different aspects of risk.
External perspectives through consultants, subject matter experts from other sites, or industry benchmarking can provide additional protection against organizational blind spots. However, as the PIC/S guidance emphasizes, these external resources should facilitate and advise rather than replace internal ownership and accountability.
Rotating team membership for ongoing risk assessment activities prevents the development of group biases and ensures fresh perspectives on familiar processes. This also supports knowledge transfer and prevents critical risk assessment capabilities from becoming concentrated in specific individuals.
Evidence-Based Analysis Requirements
Scientific justification for all risk assessment conclusions requires teams to base their analysis on objective, verifiable data rather than assumptions or intuitive judgments. This includes collecting comprehensive information about process performance, material characteristics, equipment reliability, and environmental factors before drawing conclusions about risk levels.
Assumption documentation makes implicit beliefs explicit and subject to challenge. Any assumptions made during risk assessment must be clearly identified, justified with available evidence, and flagged for future validation. This transparency helps identify areas where additional data collection may be needed and prevents assumptions from becoming accepted facts over time.
Evidence quality assessment evaluates the strength and reliability of information used to support risk assessment conclusions. This includes understanding limitations, uncertainties, and potential sources of bias in the data itself.
Structured uncertainty analysisexplicitly addresses areas where knowledge is incomplete or confidence is low. Rather than treating uncertainty as a weakness to be minimized, mature quality systems acknowledge uncertainty and design controls that remain effective despite incomplete information.
Continuous Monitoring and Reassessment Systems
Performance validation provides ongoing verification of risk assessment accuracy through operational performance data. The PIC/S guidance emphasizes that risk assessments should be “periodically reviewed for currency and effectiveness” with systems to track how well predicted risks align with actual experience.
Assumption testing uses operational data to validate or refute assumptions embedded in risk assessments. When monitoring reveals discrepancies between predicted and actual performance, this triggers systematic review of the original assessment to identify potential sources of bias or incomplete analysis.
Feedback loopsensure that lessons learned from risk assessment performance are incorporated into future assessments. This includes both successful risk predictions and instances where significant risks were initially overlooked.
Adaptive learning systems use accumulated experience to improve risk assessment methodologies and training programs. Organizations can track patterns in assessment effectiveness to identify systematic biases or knowledge gaps that require attention.
Knowledge Management as the Foundation of Cognitive Excellence
The Critical Challenge of Tacit Knowledge Capture
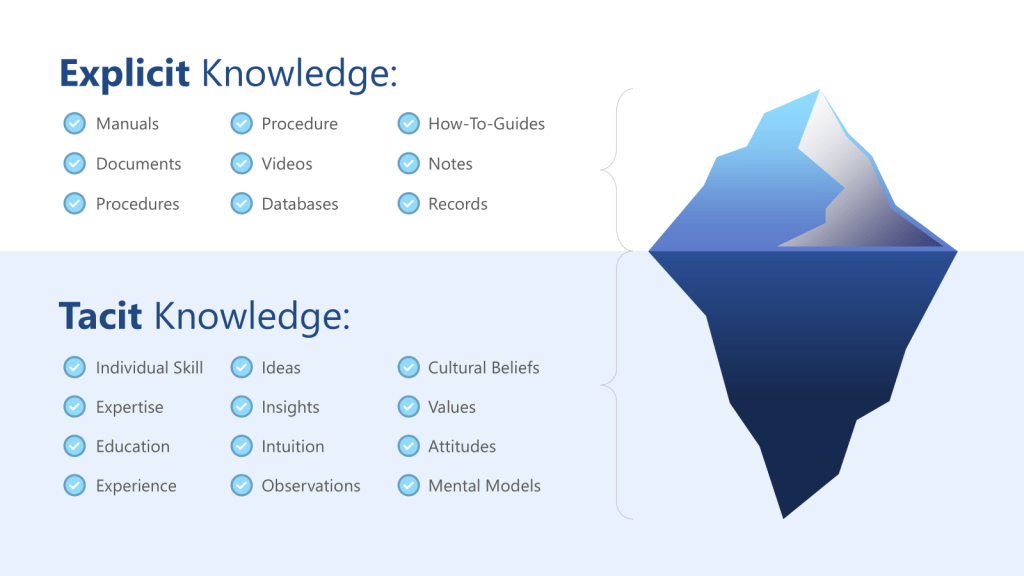
ICH Q10’s definition of knowledge management as “a systematic approach to acquiring, analysing, storing and disseminating information related to products, manufacturing processes and components” provides the regulatory framework, but the cognitive dimensions of knowledge management are equally critical. The distinction between tacit knowledge (experiential, intuitive understanding) and explicit knowledge (documented procedures and data) becomes crucial when designing systems to support effective risk assessment.
Tacit knowledge capture represents one of the most significant challenges in pharmaceutical quality systems. The experienced process engineer who can “feel” when a process is running correctly possesses invaluable knowledge, but this knowledge remains vulnerable to loss through retirements, organizational changes, or simply the passage of time. More critically, tacit knowledge often contains embedded assumptions that may become outdated as processes, materials, or environmental conditions change.
Structured knowledge elicitation processes systematically capture not just what experts know, but how they know it—the cues, patterns, and reasoning processes that guide their decision-making. This involves techniques such as cognitive interviewing, scenario-based discussions, and systematic documentation of decision rationales that make implicit knowledge explicit and subject to validation.
Knowledge validation and updating cycles ensure that captured knowledge remains current and accurate. This is particularly important for tacit knowledge, which may be based on historical conditions that no longer apply. Systematic processes for testing and updating knowledge prevent the accumulation of outdated assumptions that can compromise risk assessment effectiveness.
Expertise Distribution and Access
Knowledge networks provide systematic approaches to connecting decision-makers with relevant expertise when complex risk assessments require specialized knowledge. Rather than assuming that generalist teams can address all risk assessment challenges, mature organizations develop capabilities to identify when specialized expertise is required and ensure it is accessible when needed.
Expertise mapping creates systematic inventories of knowledge and capabilities distributed throughout the organization. This includes not just formal qualifications and roles, but understanding of specific process knowledge, problem-solving experience, and decision-making capabilities that may be relevant to risk assessment activities.
Dynamic expertise allocation ensures that appropriate knowledge is available for specific risk assessment challenges. This might involve bringing in experts from other sites for novel process assessments, engaging specialists for complex technical evaluations, or providing access to external expertise when internal capabilities are insufficient.
Knowledge accessibility systems make relevant information available at the point of decision-making through searchable databases, expert recommendation systems, and structured repositories that support rapid access to historical decisions, lessons learned, and validated approaches.
Knowledge Quality and Validation
Systematic assumption identification makes embedded beliefs explicit and subject to validation. Knowledge management systems must capture not just conclusions and procedures, but the assumptions and reasoning that support them. This enables systematic testing and updating when new evidence emerges.
Evidence-based knowledge validation uses operational performance data, scientific literature, and systematic observation to test the accuracy and currency of organizational knowledge. This includes both confirming successful applications and identifying instances where accepted knowledge may be incomplete or outdated.
Knowledge audit processes systematically evaluate the quality, completeness, and accessibility of knowledge required for effective risk assessment. This includes identifying knowledge gaps that may compromise assessment effectiveness and developing plans to address critical deficiencies.
Continuous knowledge improvement integrates lessons learned from risk assessment performance into organizational knowledge bases. When assessments prove accurate or identify overlooked risks, these experiences become part of organizational learning that improves future performance.
Integration with Risk Assessment Processes
Knowledge-enabled risk assessment systematically integrates relevant organizational knowledge into risk evaluation processes. This includes access to historical performance data, previous risk assessments for similar situations, lessons learned from comparable processes, and validated assumptions about process behaviors and control effectiveness.
Decision support integration provides risk assessment teams with structured access to relevant knowledge at each stage of the assessment process. This might include automated recommendations for relevant expertise, access to similar historical assessments, or prompts to consider specific knowledge domains that may be relevant.
Knowledge visualization and analytics help teams identify patterns, relationships, and insights that might not be apparent from individual data sources. This includes trend analysis, correlation identification, and systematic approaches to integrating information from multiple sources.
Real-time knowledge validation uses ongoing operational performance to continuously test and refine knowledge used in risk assessments. Rather than treating knowledge as static, these systems enable dynamic updating based on accumulating evidence and changing conditions.
A Maturity Model for Cognitive Excellence in Risk Management
Level 1: Reactive – The Bias-Blind Organization
Organizations at the reactive level operate with ad hoc risk assessments that rely heavily on individual judgment with minimal recognition of cognitive bias effects. Risk assessments are typically performed by whoever is available rather than teams with appropriate expertise, and conclusions are based primarily on immediate experience or intuitive responses.
Knowledge management characteristics at this level include isolated expertise with no systematic capture or sharing mechanisms. Critical knowledge exists primarily as tacit knowledge held by specific individuals, creating vulnerabilities when personnel changes occur. Documentation is minimal and typically focused on conclusions rather than reasoning processes or supporting evidence.
Cognitive bias manifestations are pervasive but unrecognized. Teams routinely fall prey to anchoring, confirmation bias, and availability bias without awareness of these influences on their conclusions. Unjustified assumptions are common and remain unchallenged because there are no systematic processes to identify or test them.
Decision-making processes lack structure and repeatability. Risk assessments may produce different conclusions when performed by different teams or at different times, even when addressing identical situations. There are no systematic approaches to ensuring comprehensive risk identification or validating assessment conclusions.
Typical challenges include recurring problems despite seemingly adequate risk assessments, inconsistent risk assessment quality across different teams or situations, and limited ability to learn from assessment experience. Organizations at this level often experience surprise failures where significant risks were not identified during formal risk assessment processes.
Level 2: Awareness – Recognizing the Problem
Organizations advancing to the awareness level demonstrate basic recognition of cognitive bias risks with inconsistent application of structured methods. There is growing understanding that human judgment limitations can affect risk assessment quality, but systematic approaches to addressing these limitations are incomplete or irregularly applied.
Knowledge management progress includes beginning attempts at knowledge documentation and expert identification. Organizations start to recognize the value of capturing expertise and may implement basic documentation requirements or expert directories. However, these efforts are often fragmented and lack systematic integration with risk assessment processes.
Cognitive bias recognition becomes more systematic, with training programs that help personnel understand common bias types and their potential effects on decision-making. However, awareness does not consistently translate into behavior change, and bias mitigation techniques are applied inconsistently across different assessment situations.
Decision-making improvements include basic templates or checklists that promote more systematic consideration of risk factors. However, these tools may be applied mechanically without deep understanding of their purpose or integration with broader quality system objectives.
Emerging capabilities include better documentation of assessment rationales, more systematic involvement of diverse perspectives in some assessments, and beginning recognition of the need for external expertise in complex situations. However, these practices are not yet embedded consistently throughout the organization.
Level 3: Systematic – Building Structured Defenses
Level 3 organizations implement standardized risk assessment protocols with built-in bias checks and documented decision rationales. There is systematic recognition that cognitive limitations require structured countermeasures, and processes are designed to promote more reliable decision-making.
Knowledge management formalization includes formal knowledge management processes including expert networks and structured knowledge capture. Organizations develop systematic approaches to identifying, documenting, and sharing expertise relevant to risk assessment activities. Knowledge is increasingly treated as a strategic asset requiring active management.
Bias mitigation integration embeds cognitive bias awareness and countermeasures into standard risk assessment procedures. This includes systematic use of devil’s advocate processes, structured approaches to challenging assumptions, and requirements for evidence-based justification of conclusions.
Structured decision processes ensure consistent application of comprehensive risk assessment methodologies with clear requirements for documentation, evidence, and review. Teams follow standardized approaches that promote systematic consideration of relevant risk factors while providing flexibility for situation-specific analysis.
Quality characteristics include more consistent risk assessment performance across different teams and situations, systematic documentation that enables effective review and learning, and better integration of risk assessment activities with broader quality system objectives.
Level 4: Integrated – Cultural Transformation
Level 4 organizations achieve cross-functional teams, systematic training, and continuous improvement processes with bias mitigation embedded in quality culture. Cognitive excellence becomes an organizational capability rather than a set of procedures, supported by culture, training, and systematic reinforcement.
Knowledge management integration fully integrates knowledge management with risk assessment processes and supports these with technology platforms. Knowledge flows seamlessly between different organizational functions and activities, with systematic approaches to maintaining currency and relevance of organizational knowledge assets.
Cultural integration creates organizational environments where systematic, evidence-based decision-making is expected and rewarded. Personnel at all levels understand the importance of cognitive rigor and actively support systematic approaches to risk assessment and decision-making.
Systematic training and development builds organizational capabilities in both technical risk assessment methodologies and cognitive skills required for effective application. Training programs address not just what tools to use, but how to think systematically about complex risk assessment challenges.
Continuous improvement mechanisms systematically analyze risk assessment performance to identify opportunities for enhancement and implement improvements in methodologies, training, and support systems.
Level 5: Optimizing – Predictive Intelligence
Organizations at the optimizing level implement predictive analytics, real-time bias detection, and adaptive systems that learn from assessment performance. These organizations leverage advanced technologies and systematic approaches to achieve exceptional performance in risk assessment and management.
Predictive capabilities enable organizations to anticipate potential risks and bias patterns before they manifest in assessment failures. This includes systematic monitoring of assessment performance, early warning systems for potential cognitive failures, and proactive adjustment of assessment approaches based on accumulated experience.
Adaptive learning systems continuously improve organizational capabilities based on performance feedback and changing conditions. These systems can identify emerging patterns in risk assessment challenges and automatically adjust methodologies, training programs, and support systems to maintain effectiveness.
Industry leadership characteristics include contributing to industry knowledge and best practices, serving as benchmarks for other organizations, and driving innovation in risk assessment methodologies and cognitive excellence approaches.
Implementation Strategies: Building Cognitive Excellence
Training and Development Programs
Cognitive bias awareness training must go beyond simple awareness to build practical skills in bias recognition and mitigation. Effective programs use case studies from pharmaceutical manufacturing to illustrate how biases can lead to serious consequences and provide hands-on practice with bias recognition and countermeasure application.
Critical thinking skill development builds capabilities in systematic analysis, evidence evaluation, and structured problem-solving. These programs help personnel recognize when situations require careful analysis rather than intuitive responses and provide tools for engaging systematic thinking processes.
Risk assessment methodology training combines technical instruction in formal risk assessment tools with cognitive skills required for effective application. This includes understanding when different methodologies are appropriate, how to adapt tools for specific situations, and how to recognize and address limitations in chosen approaches.
Knowledge management skills help personnel contribute effectively to organizational knowledge capture, validation, and sharing activities. This includes skills in documenting decision rationales, participating in knowledge networks, and using knowledge management systems effectively.
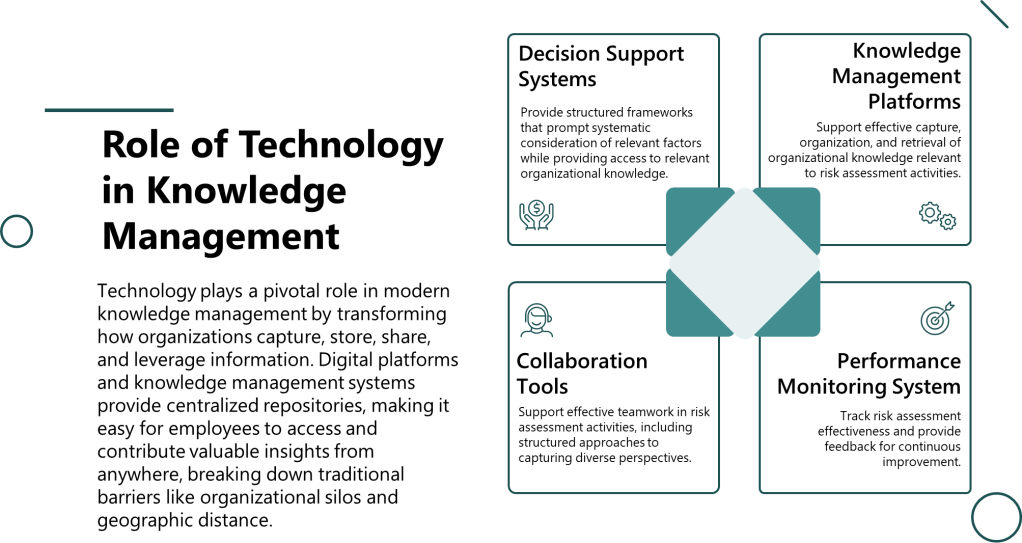
Technology Integration
Decision support systems provide structured frameworks that prompt systematic consideration of relevant factors while providing access to relevant organizational knowledge. These systems help teams engage appropriate cognitive processes while avoiding common bias traps.
Knowledge management platforms support effective capture, organization, and retrieval of organizational knowledge relevant to risk assessment activities. Advanced systems can provide intelligent recommendations for relevant expertise, historical assessments, and validated approaches based on assessment context.
Performance monitoring systems track risk assessment effectiveness and provide feedback for continuous improvement. These systems can identify patterns in assessment performance that suggest systematic biases or knowledge gaps requiring attention.
Collaboration tools support effective teamwork in risk assessment activities, including structured approaches to capturing diverse perspectives and managing group decision-making processes to avoid groupthink and other collective biases.
Organizational Culture Development
Leadership commitment demonstrates visible support for systematic, evidence-based approaches to risk assessment. This includes providing adequate time and resources for thorough analysis, recognizing effective risk assessment performance, and holding personnel accountable for systematic approaches to decision-making.
Psychological safety creates environments where personnel feel comfortable challenging assumptions, raising concerns about potential risks, and admitting uncertainty or knowledge limitations. This requires organizational cultures that treat questioning and systematic analysis as valuable contributions rather than obstacles to efficiency.
Learning orientation emphasizes continuous improvement in risk assessment capabilities rather than simply achieving compliance with requirements. Organizations with strong learning cultures systematically analyze assessment performance to identify improvement opportunities and implement enhancements in methodologies and capabilities.
Knowledge sharing cultures actively promote the capture and dissemination of expertise relevant to risk assessment activities. This includes recognition systems that reward knowledge sharing, systematic approaches to capturing lessons learned, and integration of knowledge management activities with performance evaluation and career development.
Conducting a Knowledge Audit for Risk Assessment
Organizations beginning this journey should start with a systematic knowledge audit that identifies potential vulnerabilities in expertise availability and access. This audit should address several key areas:
Expertise mapping to identify knowledge holders, their specific capabilities, and potential vulnerabilities from personnel changes or workload concentration. This includes both formal expertise documented in job descriptions and informal knowledge that may be critical for effective risk assessment.
Knowledge accessibility assessment to evaluate how effectively relevant knowledge can be accessed when needed for risk assessment activities. This includes both formal systems such as databases and informal networks that provide access to specialized expertise.
Knowledge quality evaluation to assess the currency, accuracy, and completeness of knowledge used to support risk assessment decisions. This includes identifying areas where assumptions may be outdated or where knowledge gaps may compromise assessment effectiveness.
Cognitive bias vulnerability assessment to identify situations where systematic biases are most likely to affect risk assessment conclusions. This includes analyzing past assessment performance to identify patterns that suggest bias effects and evaluating current processes for bias mitigation effectiveness.
Structured assessment protocols should incorporate specific checkpoints and requirements designed to counter known cognitive biases. This includes mandatory consideration of alternative explanations, requirements for external validation of conclusions, and systematic approaches to challenging preferred solutions.
Team composition guidelines should ensure appropriate cognitive diversity while maintaining technical competence. This includes balancing experience levels, functional backgrounds, and thinking styles to maximize the likelihood of identifying diverse perspectives on risk assessment challenges.
Evidence requirements should specify the types and quality of information required to support different types of risk assessment conclusions. This includes guidelines for evaluating evidence quality, addressing uncertainty, and documenting limitations in available information.
Review and validation processes should provide systematic quality checks on risk assessment conclusions while identifying potential bias effects. This includes independent review requirements, structured approaches to challenging conclusions, and systematic tracking of assessment performance over time.
Building Knowledge-Enabled Decision Making
Integration strategies should systematically connect knowledge management activities with risk assessment processes. This includes providing risk assessment teams with structured access to relevant organizational knowledge and ensuring that assessment conclusions contribute to organizational learning.
Technology selection should prioritize systems that enhance rather than replace human judgment while providing effective support for systematic decision-making processes. This includes careful evaluation of user interface design, integration with existing workflows, and alignment with organizational culture and capabilities.
Performance measurement should track both risk assessment effectiveness and knowledge management performance to ensure that both systems contribute effectively to organizational objectives. This includes metrics for knowledge quality, accessibility, and utilization as well as traditional risk assessment performance indicators.
Continuous improvement processes should systematically analyze performance in both risk assessment and knowledge management to identify enhancement opportunities and implement improvements in methodologies, training, and support systems.
Excellence Through Systematic Cognitive Development
The journey toward cognitive excellence in pharmaceutical risk management requires fundamental recognition that human cognitive limitations are not weaknesses to be overcome through training alone, but systematic realities that must be addressed through thoughtful system design. The PIC/S observations of unjustified assumptions, incomplete risk identification, and inappropriate tool application represent predictable patterns that emerge when sophisticated professionals operate without systematic support for cognitive excellence.
Excellence in this context means designing quality systems that work with human cognitive capabilities rather than against them. This requires integrating knowledge management principles with cognitive science insights to create environments where systematic, evidence-based decision-making becomes natural and sustainable. It means moving beyond hope that awareness will overcome bias toward systematic implementation of structures, processes, and cultures that promote cognitive rigor.
Elegance lies in recognizing that the most sophisticated risk assessment methodologies are only as effective as the cognitive processes that apply them. True elegance in quality system design comes from seamlessly integrating technical excellence with cognitive support, creating systems where the right decisions emerge naturally from the intersection of human expertise and systematic process.
Organizations that successfully implement these approaches will develop competitive advantages that extend far beyond regulatory compliance. They will build capabilities in systematic decision-making that improve performance across all aspects of pharmaceutical quality management. They will create resilient systems that can adapt to changing conditions while maintaining consistent effectiveness. Most importantly, they will develop cultures of excellence that attract and retain exceptional talent while continuously improving their capabilities.
The framework presented here provides a roadmap for this transformation, but each organization must adapt these principles to their specific context, culture, and capabilities. The maturity model offers a path for progressive development that builds capabilities systematically while delivering value at each stage of the journey.
As we face increasingly complex pharmaceutical manufacturing challenges and evolving regulatory expectations, the organizations that invest in systematic cognitive excellence will be best positioned to protect patient safety while achieving operational excellence. The choice is not whether to address these cognitive foundations of quality management, but how quickly and effectively we can build the capabilities required for sustained success in an increasingly demanding environment.
The cognitive foundations of pharmaceutical quality excellence represent both opportunity and imperative. The opportunity lies in developing systematic capabilities that transform good intentions into consistent results. The imperative comes from recognizing that patient safety depends not just on our technical knowledge and regulatory compliance, but on our ability to think clearly and systematically about complex risks in an uncertain world.
Reflective Questions for Implementation
How might you assess your organization’s current vulnerability to the three PIC/S observations in your risk management practices? What patterns in past risk assessment performance might indicate systematic cognitive biases affecting your decision-making processes?
Where does critical knowledge for risk assessment currently reside in your organization, and how accessible is it when decisions must be made? What knowledge audit approach would be most valuable for identifying vulnerabilities in your current risk management capabilities?
Which level of the cognitive bias mitigation maturity model best describes your organization’s current state, and what specific capabilities would be required to advance to the next level? How might you begin building these capabilities while maintaining current operational effectiveness?
What systematic changes in training, process design, and cultural expectations would be required to embed cognitive excellence into your quality culture? How would you measure progress in building these capabilities and demonstrate their value to organizational leadership?
ICH Q8 (Pharmaceutical Development), Q9 (Quality Risk Management), and Q10 (Pharmaceutical Quality System) provide a comprehensive framework for transforming change management from a reactive compliance exercise into a strategic enabler of quality and innovation.
The ICH Q8-Q10 triad is my favorite framework pharmaceutical quality systems: Q8’s Quality by Design (QbD) principles establish proactive identification of critical quality attributes (CQAs) and design spaces, shifting the paradigm from retrospective testing to prospective control; Q9 provides the scaffolding for risk-based decision-making, enabling organizations to prioritize resources based on severity, occurrence, and detectability of risks; and, Q10 closes the loop by embedding these concepts into a lifecycle-oriented quality system, emphasizing knowledge management and continual improvement.
These guidelines create a robust foundation for change control. Q8 ensures changes align with product and process understanding, Q9 enables risk-informed evaluation, and Q10 mandates systemic integration across the product lifecycle. This triad rejects the notion of change control as a standalone procedure, instead positioning it as a manifestation of organizational quality culture.
The PIC/S Perspective: Risk-Based Change Management
The PIC/S guidance (PI 054-1) reinforces ICH principles by offering a methodology that emphasizes effectiveness as the cornerstone of change management. It outlines four pillars:
Proposal and Impact Assessment: Systematic evaluation of cross-functional impacts, including regulatory filings, process interdependencies, and stakeholder needs.
Risk Classification: Stratifying changes as critical/major/minor based on potential effects on product quality, patient safety, and data integrity.
Implementation with Interim Controls: Bridging current and future states through mitigations like enhanced monitoring or temporary procedural adjustments.
Effectiveness Verification: Post-implementation reviews using metrics aligned with change objectives, supported by tools like statistical process control (SPC) or continued process verification (CPV).
This guidance operationalizes ICH concepts by mandating traceability from change rationale to verified outcomes, creating accountability loops that prevent “paper compliance.”
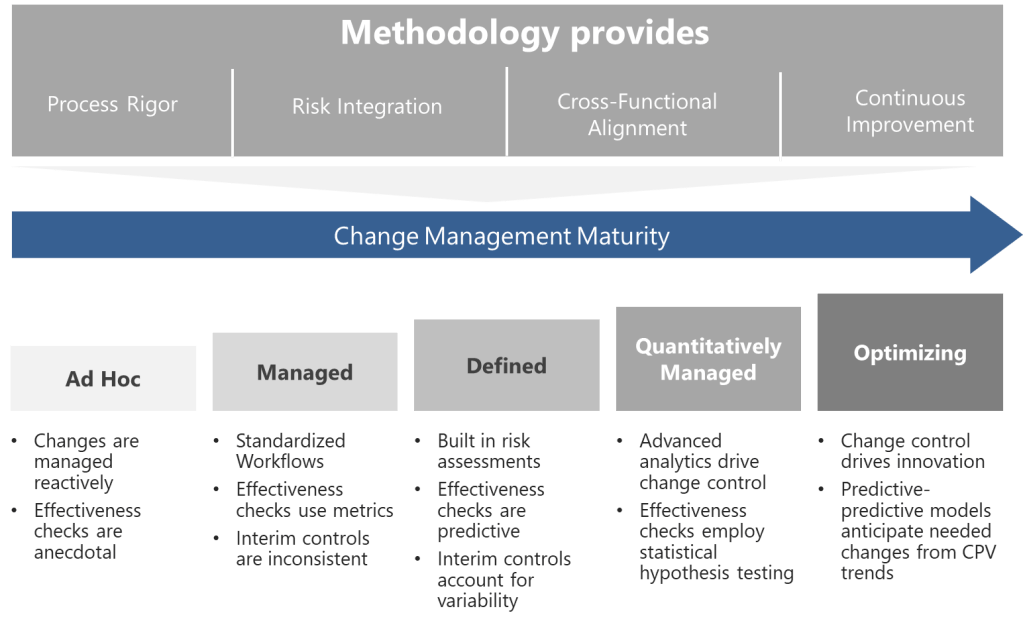
A Five-Level Maturity Model for Change Control
Building on these foundations, I propose a maturity model that evaluates organizational capability across four dimensions, each addressing critical aspects of pharmaceutical change control systems:
Process Rigor
Assesses the standardization, documentation, and predictability of change control workflows.
Higher maturity levels incorporate design space utilization (ICH Q8), automated risk thresholds, and digital tools like Monte Carlo simulations for predictive impact modeling.
Progresses from ad hoc procedures to AI-driven, self-correcting systems that preemptively identify necessary changes via CPV trends.
Risk Integration
Measures how effectively quality risk management (ICH Q9) is embedded into decision-making.
Includes risk-based classification (critical/major/minor), use of the right tool, and dynamic risk thresholds tied to process capability indices (CpK/PpK).
Evaluates collaboration between QA, regulatory, manufacturing, and supply chain teams during change evaluation.
Maturity is reflected in centralized review boards, real-time data integration (e.g., ERP/LIMS connectivity), and harmonized procedures across global sites.
Continuous Improvement
Tracks the organization’s ability to learn from past changes and innovate.
Incorporates metrics like “first-time regulatory acceptance rate” and “change-related deviation reduction.”
Top-tier organizations use post-change data to refine design spaces and update control strategies.
Level 1: Ad Hoc (Chaotic)
At this initial stage, changes are managed reactively. Procedures exist but lack standardization—departments use disparate tools, and decisions rely on individual expertise rather than systematic risk assessment. Effectiveness checks are anecdotal, often reduced to checkbox exercises. Organizations here frequently experience regulatory citations related to undocumented changes or inadequate impact assessments.
Progression Strategy: Begin by mapping all change types and aligning them with ICH Q9 risk principles. Implement a centralized change control procedure with mandatory risk classification.
Level 2: Managed (Departmental)
Changes follow standardized workflows within functions, but silos persist. Risk assessments are performed but lack cross-functional input, leading to unanticipated impacts. Effectiveness checks use basic metrics (e.g., # of changes), yet data analysis remains superficial. Interim controls are applied inconsistently, often overcompensating with excessive conservatism or being their in name only.
Progression Strategy: Establish cross-functional change review boards. Introduce the right level of formality of risk for changes and integrate CPV data into effectiveness reviews.
Level 3: Defined (Integrated)
The organization achieves horizontal integration. Changes trigger automated risk assessments using predefined criteria from ICH Q8 design spaces. Effectiveness checks leverage predictive analytics, comparing post-change performance against historical baselines. Knowledge management systems capture lessons learned, enabling proactive risk identification. Interim controls are fully operational, with clear escalation paths for unexpected variability.
Progression Strategy: Develop a unified change control platform that connects to manufacturing execution systems (MES) and laboratory information management systems (LIMS). Implement real-time dashboards for change-related KPIs.
Level 4: Quantitatively Managed (Predictive)
Advanced analytics drive change control. Machine learning models predict change impacts using historical data, reducing assessment timelines. Risk thresholds dynamically adjust based on process capability indices (CpK/PpK). Effectiveness checks employ statistical hypothesis testing, with sample sizes calculated via power analysis. Regulatory submissions for post-approval changes are partially automated through ICH Q12-enabled platforms.
Progression Strategy: Pilot digital twins for high-complexity changes, simulating outcomes before implementation. Formalize partnerships with regulators for parallel review of major changes.
Level 5: Optimizing (Self-Correcting)
Change control becomes a source of innovation. Predictive-predictive models anticipate needed changes from CPV trends. Change histories provide immutable audit trails across the product. Autonomous effectiveness checks trigger corrective actions via integrated CAPA systems. The organization contributes to industry-wide maturity through participation in various consensus standard and professional associations.
Progression Strategy: Institutionalize a “change excellence” function focused on benchmarking against emerging technologies like AI-driven root cause analysis.
Methodological Pillars: From Framework to Practice
Translating this maturity model into practice requires three methodological pillars:
1. QbD-Driven Change Design Leverage Q8’s design space concepts to predefine allowable change ranges. Changes outside the design space trigger Q9-based risk assessments, evaluating impacts on CQAs using tools like cause-effect matrices. Fully leverage Q12.
2. Risk-Based Resourcing Apply Q9’s risk prioritization to allocate resources proportionally. A minor packaging change might require a 2-hour review by QA, while a novel drug product process change engages R&D, regulatory, and supply chain teams in a multi-week analysis. Remember, the “level of effort commensurate with risk” prevents over- or under-management.
3. Closed-Loop Verification Align effectiveness checks with Q10’s lifecycle approach. Post-change monitoring periods are determined by statistical confidence levels rather than fixed durations. For instance, a formulation change might require 10 consecutive batches within CpK >1.33 before closure. PIC/S-mandated evaluations of unintended consequences are automated through anomaly detection algorithms.
Overcoming Implementation Barriers
Cultural and technical challenges abound in maturity progression. Common pitfalls include:
Overautomation: Implementing digital tools before standardizing processes, leading to “garbage in, gospel out” scenarios.
Patient-Centric Changes: Direct integration of patient-reported outcomes (PROs) into change effectiveness criteria.
Maturity as a Journey, Not a Destination
The proposed model provides a roadmap—not a rigid prescription—for advancing change control. By grounding progression in ICH Q8-Q10 and PIC/S principles, organizations can systematically enhance their change agility while maintaining compliance. Success requires viewing maturity not as a compliance milestone but as a cultural commitment to excellence, where every change becomes an opportunity to strengthen quality and accelerate innovation.
In an era of personalized medicines and decentralized manufacturing, the ability to manage change effectively will separate thriving organizations from those merely surviving. The journey begins with honest self-assessment against this model and a willingness to invest in the systems, skills, and culture that make maturity possible.
The strategic utilization of supplier documentation in qualification processes presents a significant opportunity to enhance efficiency while maintaining strict quality standards. Determining what supplier documentation can be accepted and what aspects require additional qualification is critical for streamlining validation activities without compromising product quality or patient safety.
Regulatory Framework Supporting Supplier Documentation Use
Regulatory bodies increasingly recognize the value of leveraging third-party documentation when properly evaluated and integrated into qualification programs. The FDA’s 2011 Process Validation Guidance embraces risk-based approaches that focus resources on critical aspects rather than duplicating standard testing. This guidance references the ASTM E2500 standard, which explicitly addresses the use of supplier documentation in qualification activities.
The EU GMP Annex 15 provides clear regulatory support, stating: “Data supporting qualification and/or validation studies which were obtained from sources outside of the manufacturers own programmes may be used provided that this approach has been justified and that there is adequate assurance that controls were in place throughout the acquisition of such data.” This statement offers a regulatory pathway for incorporating supplier documentation, provided proper controls and justification exist.
ICH Q9 further supports this approach by encouraging risk-based allocation of resources, allowing companies to focus qualification efforts on areas of highest risk while leveraging supplier documentation for well-controlled, lower-risk aspects. The integration of these regulatory perspectives creates a framework that enables efficient qualification strategies while maintaining regulatory compliance.
Benefits of Utilizing Supplier Documentation in Qualification
Biotech manufacturing systems present unique challenges due to their complexity, specialized nature, and biological processes. Leveraging supplier documentation offers multiple advantages in this context:
Supplier expertise in specialized biotech equipment often exceeds that available within pharmaceutical companies. This expertise encompasses deep understanding of complex technologies such as bioreactors, chromatography systems, and filtration platforms that represent years of development and refinement. Manufacturers of bioprocess equipment typically employ specialists who design and test equipment under controlled conditions unavailable to end users.
Integration of engineering documentation into qualification protocols can reduce project timelines, while significantly decreasing costs associated with redundant testing. This efficiency is particularly valuable in biotech, where manufacturing systems frequently incorporate numerous integrated components from different suppliers.
By focusing qualification resources on truly critical aspects rather than duplicating standard supplier testing, organizations can direct expertise toward product-specific challenges and integration issues unique to their manufacturing environment. This enables deeper verification of critical aspects that directly impact product quality rather than dispersing resources across standard equipment functionality tests.
Criteria for Acceptable Supplier Documentation
Audit of the Supplier
Supplier Quality System Assessment
Before accepting any supplier documentation, a thorough assessment of the supplier’s quality system must be conducted. This assessment should evaluate the following specific elements:
Quality management systems certification to relevant standards with verification of certification scope and validity. This should include review of recent certification audit reports and any major findings.
Document control systems that demonstrate proper version control, appropriate approvals, secure storage, and systematic review and update cycles. Specific attention should be paid to engineering document management systems and change control procedures for technical documentation.
Training programs with documented evidence of personnel qualification, including training matrices showing alignment between job functions and required training. Training records should demonstrate both initial training and periodic refresher training, particularly for personnel involved in critical testing activities.
Change control processes with formal impact assessments, appropriate review levels, and implementation verification. These processes should specifically address how changes to equipment design, software, or testing protocols are managed and documented.
Deviation management systems with documented root cause analysis, corrective and preventive actions, and effectiveness verification. The system should demonstrate formal investigation of testing anomalies and resolution of identified issues prior to completion of supplier testing.
Test equipment calibration and maintenance programs with NIST-traceable standards, appropriate calibration frequencies, and out-of-tolerance investigations. Records should demonstrate that all test equipment used in generating qualification data was properly calibrated at the time of testing.
Software validation practices aligned with GAMP5 principles, including risk-based validation approaches for any computer systems used in equipment testing or data management. This should include validation documentation for any automated test equipment or data acquisition systems.
Internal audit processes with independent auditors, documented findings, and demonstrable follow-up actions. Evidence should exist that the supplier conducts regular internal quality audits of departments involved in equipment design, manufacturing, and testing.
Technical Capability Verification
Supplier technical capability must be verified through:
Documentation of relevant experience with similar biotech systems, including a portfolio of comparable projects successfully completed. This should include reference installations at regulated pharmaceutical or biotech companies with complexity similar to the proposed equipment.
Technical expertise of key personnel demonstrated through formal qualifications, industry experience, and specific expertise in biotech applications. Review should include CVs of key personnel who will be involved in equipment design, testing, and documentation.
Testing methodologies that incorporate scientific principles, appropriate statistics, and risk-based approaches. Documentation should demonstrate test method development with sound scientific rationales and appropriate controls.
Calibrated and qualified test equipment with documented measurement uncertainties appropriate for the parameters being measured. This includes verification that measurement capabilities exceed the required precision for critical parameters by an appropriate margin.
GMP understanding demonstrated through documented training, experience in regulated environments, and alignment of test protocols with GMP principles. Personnel should demonstrate awareness of regulatory requirements specific to biotech applications.
Measurement traceability to national standards with documented calibration chains for all critical measurements. This should include identification of reference standards used and their calibration status.
Design control processes aligned with recognized standards including design input review, risk analysis, design verification, and design validation. Design history files should be available for review to verify systematic development approaches.
Documentation Quality Requirements
Acceptable supplier documentation must demonstrate:
Creation under GMP-compliant conditions with evidence of training for personnel generating the documentation. Records should demonstrate that personnel had appropriate training in documentation practices and understood the criticality of accurate data recording.
Compliance with GMP documentation practices including contemporaneous recording, no backdating, proper error correction, and use of permanent records. Documents should be reviewed for evidence of proper data recording practices such as signed and dated entries, proper correction of errors, and absence of unexplained gaps.
Completeness with clearly defined acceptance criteria established prior to testing. Pre-approved protocols should define all test parameters, conditions, and acceptance criteria without post-testing modifications.
Actual test results rather than summary statements, with raw data supporting reported values. Testing documentation should include actual measured values, not just pass/fail determinations, and should provide sufficient detail to allow independent evaluation.
Deviation records with thorough investigations and appropriate resolutions. Any testing anomalies should be documented with formal investigations, root cause analysis, and justification for any retesting or data exclusion.
Traceability to requirements through clear linkage between test procedures and equipment specifications. Each test should reference the specific requirement or specification it is designed to verify.
Authorization by responsible personnel with appropriate signatures and dates. Documents should demonstrate review and approval by qualified individuals with defined responsibilities in the testing process.
Data integrity controls including audit trails for electronic data, validated computer systems, and measures to prevent unauthorized modification. Evidence should exist that data security measures were in place during testing and documentation generation.
Statistical analysis and justification where appropriate, particularly for performance data involving multiple measurements or test runs. Where sampling is used, justification for sample size and statistical power should be provided.
Adherence to established industry standards and design codes relevant to biotech equipment. This includes documentation citing specific standards applied during design and evidence of compliance verification.
Implementation of systematic design methodologies including requirements gathering, conceptual design, detailed design, and design review phases. Design documentation should demonstrate progression through formal design stages with appropriate approvals at each stage.
Application of appropriate testing protocols based on equipment type, criticality, and intended use. Testing strategies should be aligned with industry norms for similar equipment and demonstrate appropriate rigor.
Maintenance of equipment calibration throughout testing phases with records demonstrating calibration status. All test equipment should be documented as calibrated before and after critical testing activities.
Documentation accuracy and completeness demonstrated through systematic review processes and quality checks. Evidence should exist of multiple review levels for critical documentation and formal approval processes.
Implementation of appropriate commissioning procedures aligned with recognized industry practices. Commissioning plans should demonstrate systematic verification of all equipment functions and utilities.
Formal knowledge transfer processes ensuring proper communication between design, manufacturing, and qualification teams. Evidence should exist of structured handover meetings or documentation between project phases.
Types of Supplier Documentation That Can Be Leveraged
When the above criteria are met, the following specific types of supplier documentation can potentially be leveraged.
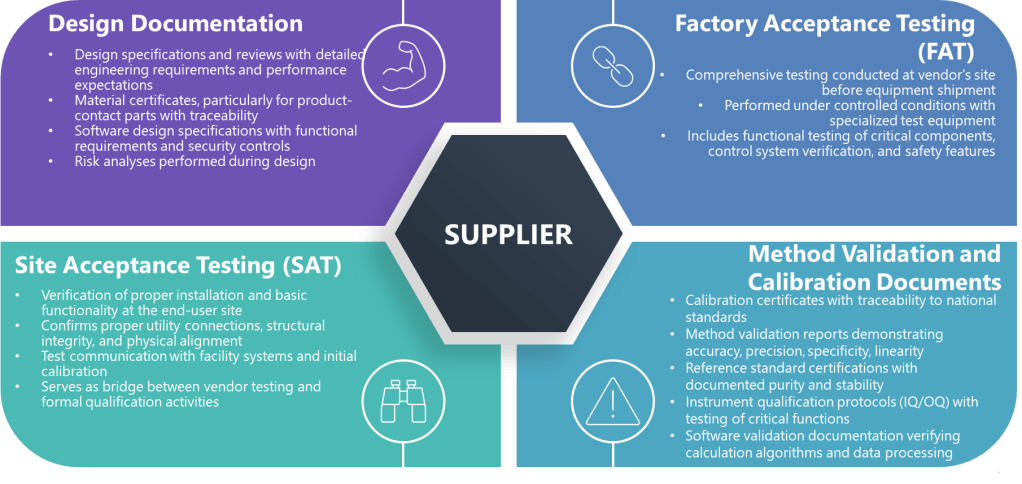
Factory Acceptance Testing (FAT)
FAT documentation represents comprehensive testing at the supplier’s site before equipment shipment. These documents are particularly valuable because they often represent testing under more controlled conditions than possible at the installation site. For biotech applications, FAT documentation may include:
Functional testing of critical components with detailed test procedures, actual measurements, and predetermined acceptance criteria. This should include verification of all critical operating parameters under various operating conditions.
Control system verification through systematic testing of all control loops, alarms, and safety interlocks. Testing should demonstrate proper response to normal operating conditions as well as fault scenarios.
Material compatibility confirmation with certificates of conformance for product-contact materials and testing to verify absence of leachables or extractables that could impact product quality.
Cleaning system performance verification through spray pattern testing, coverage verification, and drainage evaluation. For CIP (Clean-in-Place) systems, this should include documented evidence of cleaning effectiveness.
Performance verification under load conditions that simulate actual production requirements, with test loads approximating actual product characteristics where possible.
Alarm and safety feature testing with verification of proper operation of all safety interlocks, emergency stops, and containment features critical to product quality and operator safety.
Software functionality testing with documented verification of all user requirements related to automation, control systems, and data management capabilities.
Site Acceptance Testing (SAT)
SAT documentation verifies proper installation and basic functionality at the end-user site. For biotech equipment, this might include:
Installation verification confirming proper utilities connections, structural integrity, and physical alignment according to engineering specifications. This should include verification of spatial requirements and accessibility for operation and maintenance.
Basic functionality testing demonstrating that all primary equipment functions operate as designed after transportation and installation. Tests should verify that no damage occurred during shipping and installation.
Communication with facility systems verification, including integration with building management systems, data historians, and centralized control systems. Testing should confirm proper data transfer and command execution between systems.
Initial calibration verification for all critical instruments and control elements, with documented evidence of calibration accuracy and stability.
Software configuration verification showing proper installation of control software, correct parameter settings, and appropriate security configurations.
Environmental conditions verification confirming that the installed location meets requirements for temperature, humidity, vibration, and other environmental factors that could impact equipment performance.
Design Documentation
Design documents that can support qualification include:
Design specifications with detailed engineering requirements, operating parameters, and performance expectations. These should include rationales for critical design decisions and risk assessments supporting design choices.
Material certificates, particularly for product-contact parts, with full traceability to raw material sources and manufacturing processes. Documentation should include testing for biocompatibility where applicable.
Software design specifications with detailed functional requirements, system architecture, and security controls. These should demonstrate structured development approaches with appropriate verification activities.
Risk analyses performed during design, including FMEA (Failure Mode and Effects Analysis) or similar systematic evaluations of potential failure modes and their impacts on product quality and safety.
Design reviews and approvals with documented participation of subject matter experts across relevant disciplines including engineering, quality, manufacturing, and validation.
Finite element analysis reports or other engineering studies supporting critical design aspects such as pressure boundaries, mixing efficiency, or temperature distribution.
Method Validation and Calibration Documents
For analytical instruments and measurement systems, supplier documentation might include:
Calibration certificates with traceability to national standards, documented measurement uncertainties, and verification of calibration accuracy across the operating range.
Method validation reports demonstrating accuracy, precision, specificity, linearity, and robustness for analytical methods intended for use with the equipment.
Reference standard certifications with documented purity, stability, and traceability to compendial standards where applicable.
Instrument qualification protocols (IQ/OQ) with comprehensive testing of all critical functions and performance parameters against predetermined acceptance criteria.
Software validation documentation showing systematic verification of all calculation algorithms, data processing functions, and reporting capabilities.
What Must Still Be Qualified By The End User
Despite the value of supplier documentation, certain aspects always require direct qualification by the end user. These areas should be the focus of end-user qualification activities:
Facility utility connections and performance verification under actual operating conditions. This must include verification that utilities (water, steam, gases, electricity) meet the required specifications at the point of use, not just at the utility generation source.
Integration with other manufacturing systems, particularly verification of interfaces between equipment from different suppliers. Testing should verify proper data exchange, sequence control, and coordinated operation during normal production and exception scenarios.
Facility-specific environmental conditions including temperature mapping, particulate monitoring, and pressure differentials that could impact biotech processes. Testing should verify that environmental conditions remain within acceptable limits during worst-case operating scenarios.
Network connectivity and data transfer verification, including security controls, backup systems, and disaster recovery capabilities. Testing should demonstrate reliable performance under peak load conditions and proper handling of network interruptions.
Alarm systems integration with central monitoring and response protocols, including verification of proper notification pathways and escalation procedures. Testing should confirm appropriate alarm prioritization and notification of responsible personnel.
Building management system interfaces with verification of environmental monitoring and control capabilities critical to product quality. Testing should verify proper feedback control and response to excursions.
Process-specific parameters beyond standard equipment functionality, with testing under actual operating conditions using representative materials. Testing should verify equipment performance with actual process materials, not just test substances.
Custom configurations for specific products, including verification of specialized equipment settings, program parameters, or mechanical adjustments unique to the user’s products.
Production-scale performance verification, with particular attention to scale-dependent parameters such as mixing efficiency, heat transfer, and mass transfer. Testing should verify that performance characteristics demonstrated at supplier facilities translate to full-scale production.
Process-specific cleaning verification, including worst-case residue removal studies and cleaning cycle development specific to the user’s products. Testing should demonstrate effective cleaning of all product-contact surfaces with actual product residues.
Specific operating ranges for the user’s process, with verification of performance at the extremes of normal operating parameters. Testing should verify capability to maintain critical parameters within required tolerances throughout production cycles.
Process-specific automation sequences and recipes with verification of all production scenarios, including exception handling and recovery procedures. Testing should verify all process recipes and automated sequences with actual production materials.
Hold time verification for intermediate process steps specific to the user’s manufacturing process. Testing should confirm product stability during maximum expected hold times between process steps.
Critical Quality Attributes
Testing related directly to product-specific critical quality attributes should generally not be delegated solely to supplier documentation, particularly for:
Bioburden and endotoxin control verification using the actual production process and materials. Testing should verify absence of microbial contamination and endotoxin introduction throughout the manufacturing process.
Product contact material compatibility studies with the specific products and materials used in production. Testing should verify absence of leachables, extractables, or product degradation due to contact with equipment surfaces.
Product-specific recovery rates and process yields based on actual production experience. Testing should verify consistency of product recovery across multiple batches and operating conditions.
Process-specific impurity profiles with verification that equipment design and operation do not introduce or magnify impurities. Testing should confirm that impurity clearance mechanisms function as expected with actual production materials.
Sterility assurance measures specific to the user’s aseptic processing approaches. Testing should verify the effectiveness of sterilization methods and aseptic techniques with the actual equipment configuration and operating procedures.
Product stability during processing with verification that equipment operation does not negatively impact critical quality attributes. Testing should confirm that product quality parameters remain within acceptable limits throughout the manufacturing process.
Process-specific viral clearance capacity for biological manufacturing processes. Testing should verify effective viral removal or inactivation capabilities with the specific operating parameters used in production.
Operational and Procedural Integration
A critical area often overlooked in qualification plans is operational and procedural integration, which requires end-user qualification for:
Operator interface verification with confirmation that user interactions with equipment controls are intuitive, error-resistant, and aligned with standard operating procedures. Testing should verify that operators can effectively control the equipment under normal and exception conditions.
Procedural workflow integration ensuring that equipment operation aligns with established manufacturing procedures and documentation systems. Testing should verify compatibility between equipment operation and procedural requirements.
Training effectiveness verification for operators, maintenance personnel, and quality oversight staff. Assessment should confirm that personnel can effectively operate, maintain, and monitor equipment in compliance with established procedures.
Maintenance accessibility and procedural verification to ensure that preventive maintenance can be performed effectively without compromising product quality. Testing should verify that maintenance activities can be performed as specified in supplier documentation.
Sampling accessibility and technique verification to ensure representative samples can be obtained safely without compromising product quality. Testing should confirm that sampling points are accessible and provide representative samples.
Change management procedures specific to the user’s quality system, with verification that equipment changes can be properly evaluated, implemented, and documented. Testing should confirm integration with the user’s change control system.
Implementing a Risk-Based Approach to Supplier Documentation
A systematic risk-based approach should be implemented to determine what supplier documentation can be leveraged and what requires additional verification:
Perform impact assessment to categorize system components based on their potential impact on product quality:
Direct impact components with immediate influence on critical quality attributes
Indirect impact components that support direct impact systems
No impact components without reasonable influence on product quality
Conduct risk analysis using formal tools such as FMEA to identify:
Critical components and functions requiring thorough qualification
Identify gaps between supplier documentation and qualification requirements by:
Mapping supplier testing to user requirements
Evaluating the quality and completeness of supplier testing
Identifying areas where supplier testing does not address user-specific requirements
Assessing the reliability and applicability of supplier data to the user’s specific application
Create targeted verification plans to address:
High-risk areas not adequately covered by supplier documentation
User-specific requirements not addressed in supplier testing
Integration points between supplier equipment and user systems
Process-specific performance requirements
This risk-based methodology ensures that qualification resources are focused on areas of highest concern while leveraging reliable supplier documentation for well-controlled aspects.
Documentation and Justification Requirements
When using supplier documentation in qualification, proper documentation and justification are essential:
Create a formal supplier assessment report documenting:
Evaluation methodology and criteria used to assess the supplier
Evidence of supplier quality system effectiveness
Verification of supplier technical capabilities
Assessment of documentation quality and completeness
Identification of any deficiencies and their resolution
Develop a gap assessment identifying:
Areas where supplier documentation meets qualification requirements
Areas requiring additional end-user verification
Rationale for decisions on accepting or supplementing supplier documentation
Risk-based justification for the scope of end-user qualification activities
Prepare a traceability matrix showing:
Mapping between user requirements and testing activities
Source of verification for each requirement (supplier or end-user testing)
Evidence of test completion and acceptance
Cross-references to specific documentation supporting requirement verification
Maintain formal acceptance of supplier documentation with:
Quality unit review and approval of supplier documentation
Documentation of any additional verification activities performed
Records of any deficiencies identified and their resolution
Evidence of conformance to predetermined acceptance criteria
Document rationale for accepting supplier documentation:
Risk-based justification for leveraging supplier testing
Assessment of supplier documentation reliability and completeness
Evaluation of supplier testing conditions and their applicability
Formal incorporation of supplier documentation into the quality system
Version control and change management for supplier documentation
Secure storage and retrieval systems for qualification records
Maintenance of complete documentation packages supporting qualification decisions
Biotech-Specific Considerations
For Cell Culture Systems:
While basic temperature, pressure, and mixing capabilities may be verified through supplier testing, product-specific parameters require end-user verification. These include:
Cell viability and growth characteristics with the specific cell lines used in production. End-user testing should verify consistent cell growth, viability, and productivity under normal operating conditions.
Metabolic profiles and nutrient consumption rates specific to the production process. Testing should confirm that equipment design supports appropriate nutrient delivery and waste removal for optimal cell performance.
Homogeneity studies for bioreactors under process-specific conditions including actual media formulations, cell densities, and production phase operating parameters. Testing should verify uniform conditions throughout the bioreactor volume during all production phases.
Cell culture monitoring systems calibration and performance with actual production cell lines and media. Testing should confirm reliable and accurate monitoring of critical culture parameters throughout the production cycle.
Scale-up effects specific to the user’s cell culture process, with verification that performance characteristics demonstrated at smaller scales translate to production scale. Testing should verify comparable cell growth kinetics and product quality across scales.
For Purification Systems
Chromatography system pressure capabilities and gradient formation may be accepted from supplier testing, but product-specific performance requires end-user verification:
Product-specific recovery, impurity clearance, and yield verification using actual production materials. Testing should confirm consistent product recovery and impurity removal across multiple cycles.
Resin lifetime and performance stability with the specific products and buffer systems used in production. Testing should verify consistent performance throughout the expected resin lifetime.
Cleaning and sanitization effectiveness specific to the user’s products and contaminants. Testing should confirm complete removal of product residues and effective sanitization between production cycles.
Column packing reproducibility and performance with production-scale columns and actual resins. Testing should verify consistent column performance across multiple packing cycles.
Buffer preparation and delivery system performance with actual buffer formulations. Testing should confirm accurate preparation and delivery of all process buffers under production conditions.
For Analytical Methods
Basic instrument functionality can be verified through supplier IQ/OQ documentation, but method-specific performance requires end-user verification:
Method-specific performance with actual product samples, including verification of specificity, accuracy, and precision with the user’s products. Testing should confirm reliable analytical performance with actual production materials.
Method robustness under the specific laboratory conditions where testing will be performed. Testing should verify consistent method performance across the range of expected operating conditions.
Method suitability for the intended use, including capability to detect relevant product variants and impurities. Testing should confirm that the method can reliably distinguish between acceptable and unacceptable product quality.
Operator technique verification to ensure consistent method execution by all analysts who will perform the testing. Assessment should confirm that all analysts can execute the method with acceptable precision and accuracy.
Data processing and reporting verification with the user’s specific laboratory information management systems. Testing should confirm accurate data transfer, calculations, and reporting.
Practical Examples
Example 1: Bioreactor Qualification
For a 2000L bioreactor system, supplier documentation might be leveraged for:
Acceptable with minimal verification: Pressure vessel certification, welding documentation, motor specification verification, basic control system functionality, standard safety features. These aspects are governed by well-established engineering standards and can be reliably verified by the supplier in a controlled environment.
Acceptable with targeted verification: Temperature control system performance, basic mixing capability, sensor calibration procedures. While these aspects can be largely verified by the supplier, targeted verification in the user’s facility ensures that performance meets process-specific requirements.
Requiring end-user qualification: Process-specific mixing studies with actual media, cell culture growth performance, specific gas transfer rates, cleaning validation with product residues. These aspects are highly dependent on the specific process and materials used and cannot be adequately verified by the supplier.
In all cases, the acceptance of supplier documentation must be documented well and performed according to GMPs and at appropriately described in the Validation Plan or other appropriate testing rationale document.
Example 2: Chromatography System Qualification
For a multi-column chromatography system, supplier documentation might be leveraged as follows:
Acceptable with minimal verification: Pressure testing of flow paths, pump performance specifications, UV detector linearity, conductivity sensor calibration, valve switching accuracy. These aspects involve standard equipment functionality that can be reliably verified by the supplier using standardized testing protocols.
Acceptable with targeted verification: Gradient formation accuracy, column switching precision, UV detection sensitivity with representative proteins, system cleaning procedures. These aspects require verification with materials similar to those used in production but can largely be addressed through supplier testing with appropriate controls.
Requiring end-user qualification: Product-specific binding capacity, elution conditions optimization, product recovery rates, impurity clearance, resin lifetime with actual process streams, cleaning validation with actual product residues. These aspects are highly process-specific and require testing with actual production materials under normal operating conditions.
The qualification approach must balance efficiency with appropriate rigor, focusing end-user testing on aspects that are process-specific or critical to product quality.
Example 3: Automated Analytical Testing System Qualification
For an automated high-throughput analytical testing platform used for product release testing, supplier documentation might be leveraged as follows:
Acceptable with minimal verification: Mechanical subsystem functionality, basic software functionality, standard instrument calibration, electrical safety features, standard data backup systems. These fundamental aspects of system performance can be reliably verified by the supplier using standardized testing protocols.
Acceptable with targeted verification: Sample throughput rates, basic method execution, standard curve generation, basic system suitability testing, data export functions. These aspects require verification with representative materials but can largely be addressed through supplier testing with appropriate controls.
Requiring end-user qualification: Method-specific performance with actual product samples, detection of product-specific impurities, method robustness under laboratory-specific conditions, integration with laboratory information management systems, data integrity controls specific to the user’s quality system, analyst training effectiveness. These aspects are highly dependent on the specific analytical methods, products, and laboratory environment.
For analytical systems involved in release testing, additional considerations include:
Verification of method transfer from development to quality control laboratories
Demonstration of consistent performance across multiple analysts
Confirmation of data integrity throughout the complete testing process
Integration with the laboratory’s sample management and result reporting systems
Alignment with regulatory filing commitments for analytical methods
This qualification strategy ensures that standard instrument functionality is efficiently verified through supplier documentation while focusing end-user resources on the product-specific aspects critical to reliable analytical results.
Conclusion: Best Practices for Supplier Documentation in Biotech Qualification
To maximize the benefits of supplier documentation while ensuring regulatory compliance in biotech qualification:
Develop clear supplier requirements early in the procurement process, with specific documentation expectations communicated before equipment design and manufacturing. These requirements should specifically address documentation format, content, and quality standards.
Establish formal supplier assessment processes with clear criteria aligned with regulatory expectations and internal quality standards. These assessments should be performed by multidisciplinary teams including quality, engineering, and manufacturing representatives.
Implement quality agreements with key equipment suppliers, explicitly defining responsibilities for documentation, testing, and qualification activities. These agreements should include specifics on documentation standards, testing protocols, and data integrity requirements.
Create standardized processes for reviewing and accepting supplier documentation based on criticality and risk assessment. These processes should include formal gap analysis and identification of supplemental testing requirements.
Apply risk-based approaches consistently when determining what can be leveraged, focusing qualification resources on aspects with highest potential impact on product quality. Risk assessments should be documented with clear rationales for acceptance decisions.
Document rationale thoroughly for acceptance decisions, including scientific justification and regulatory considerations. Documentation should demonstrate a systematic evaluation process with appropriate quality oversight.
Maintain appropriate quality oversight throughout the process, with quality unit involvement in key decisions regarding supplier documentation acceptance. Quality representatives should review and approve supplier assessment reports and qualification plans.
Implement verification activities targeting gaps and high-risk areas identified during document review, focusing on process-specific and integration aspects. Verification testing should be designed to complement, not duplicate, supplier testing.
Integrate supplier documentation within your qualification lifecycle approach, establishing clear linkages between supplier testing and overall qualification requirements. Traceability matrices should demonstrate how supplier documentation contributes to meeting qualification requirements.
The key is finding the right balance between leveraging supplier expertise and maintaining appropriate end-user verification of critical aspects that impact product quality and patient safety. Proper evaluation and integration of supplier documentation represents a significant opportunity to enhance qualification efficiency while maintaining the rigorous standards essential for biotech products. With clear criteria for acceptance, systematic risk assessment, and thorough documentation, organizations can confidently leverage supplier documentation as part of a comprehensive qualification strategy aligned with current regulatory expectations and quality best practices.
In a previous post, I discussed how overcoming subjectivity in risk management and decision-making requires fostering a culture of quality and excellence. This is an issue that it is important to continue to evaluate and push for additional improvement.
The revised ICH Q9(R1) guideline, finalized in January 2023, introduces critical updates to Quality Risk Management (QRM) practices, emphasizing the need to address subjectivity, enhance formality, improve risk-based decision-making, and manage product availability risks. These revisions aim to ensure that QRM processes are more science-driven, knowledge-based, and effective in safeguarding product quality and patient safety. Two years later it is important to continue to build on key strategies for reducing subjectivity in QRM and aligning with the updated requirements.
Understanding Subjectivity in QRM
Subjectivity in QRM arises from personal opinions, biases, heuristics, or inconsistent interpretations of risks by stakeholders. This can impact every stage of the QRM process—from hazard identification to risk evaluation and mitigation. The revised ICH Q9(R1) explicitly addresses this issue by introducing a new subsection, “Managing and Minimizing Subjectivity,” which emphasizes that while subjectivity cannot be entirely eliminated, it can be controlled through structured approaches.
The guideline highlights that subjectivity often stems from poorly designed scoring systems, differing perceptions of hazards and risks among stakeholders, and cognitive biases. To mitigate these challenges, organizations must adopt robust strategies that prioritize scientific knowledge and data-driven decision-making.
Strategies to Reduce Subjectivity
Leveraging Knowledge Management
ICH Q9(R1) underscores the importance of knowledge management as a tool to reduce uncertainty and subjectivity in risk assessments. Effective knowledge management involves systematically capturing, organizing, and applying internal and external knowledge to inform QRM activities. This includes maintaining centralized repositories for technical data, fostering real-time information sharing across teams, and learning from past experiences through structured lessons-learned processes.
By integrating knowledge management into QRM, organizations can ensure that decisions are based on comprehensive data rather than subjective estimations. For example, using historical data on process performance or supplier reliability can provide objective insights into potential risks.
To integrate knowledge management (KM) more effectively into quality risk management (QRM), organizations can implement several strategies to ensure decisions are based on comprehensive data rather than subjective estimations:
Establish Robust Knowledge Repositories
Create centralized, easily accessible repositories for storing and organizing historical data, lessons learned, and best practices. These repositories should include:
Process performance data
Supplier reliability metrics
Deviation and CAPA records
Audit findings and inspection observations
Technology transfer documentation
By maintaining these repositories, organizations can quickly access relevant historical information when conducting risk assessments.
Implement Knowledge Mapping
Conduct knowledge mapping exercises to identify key sources of knowledge within the organization. This process helps to:
The revised guideline introduces a dedicated section on risk-based decision-making, emphasizing the need for structured approaches that consider the complexity, uncertainty, and importance of decisions. Organizations should establish clear criteria for decision-making processes, define acceptable risk tolerance levels, and use evidence-based methods to evaluate options.
Structured decision-making tools can help standardize how risks are assessed and prioritized. Additionally, calibrating expert opinions through formal elicitation techniques can further reduce variability in judgments.
Addressing Cognitive Biases
Cognitive biases—such as overconfidence or anchoring—can distort risk assessments and lead to inconsistent outcomes. To address this, organizations should provide training on recognizing common biases and their impact on decision-making. Encouraging diverse perspectives within risk assessment teams can also help counteract individual biases.
For example, using cross-functional teams ensures that different viewpoints are considered when evaluating risks, leading to more balanced assessments. Regularly reviewing risk assessment outputs for signs of bias or inconsistencies can further enhance objectivity.
Enhancing Formality in QRM
ICH Q9(R1) introduces the concept of a “formality continuum,” which aligns the level of effort and documentation with the complexity and significance of the risk being managed. This approach allows organizations to allocate resources effectively by applying less formal methods to lower-risk issues while reserving rigorous processes for high-risk scenarios.
For instance, routine quality checks may require minimal documentation compared to a comprehensive risk assessment for introducing new manufacturing technologies. By tailoring formality levels appropriately, organizations can ensure consistency while avoiding unnecessary complexity.
Calibrating Expert Opinions
We need to recognize the importance of expert knowledge in QRM activities, but also acknowledges the potential for subjectivity and bias in expert judgments. We need to ensure we:
Implement formal processes for expert opinion elicitation
Use techniques to calibrate expert judgments, especially when estimating probabilities
Provide training on common cognitive biases and their impact on risk assessment
Employ diverse teams to counteract individual biases
Regularly review risk assessment outputs for signs of bias or inconsistencies
Calibration techniques may include:
Structured elicitation protocols that break down complex judgments into more manageable components
Feedback and training to help experts align their subjective probability estimates with actual frequencies of events
Using multiple experts and aggregating their judgments through methods like Cooke’s classical model
Employing facilitation techniques to mitigate groupthink and encourage independent thinking
By calibrating expert opinions, organizations can leverage valuable expertise while minimizing subjectivity in risk assessments.
Utilizing Cooke’s Classical Model
Cooke’s Classical Model is a rigorous method for evaluating and combining expert judgments to quantify uncertainty. Here are the key steps for using the Classical Model to evaluate expert judgment:
Select and calibrate experts:
Choose 5-10 experts in the relevant field
Have experts assess uncertain quantities (“calibration questions”) for which true values are known or will be known soon
These calibration questions should be from the experts’ domain of expertise
Elicit expert assessments:
Have experts provide probabilistic assessments (usually 5%, 50%, and 95% quantiles) for both calibration questions and questions of interest
Document experts’ reasoning and rationales
Score expert performance:
Evaluate experts on two measures: a) Statistical accuracy: How well their probabilistic assessments match the true values of calibration questions b) Informativeness: How precise and focused their uncertainty ranges are
Calculate performance-based weights:
Derive weights for each expert based on their statistical accuracy and informativeness scores
Experts performing poorly on calibration questions receive little or no weight
Combine expert assessments:
Use the performance-based weights to aggregate experts’ judgments on the questions of interest
This creates a “Decision Maker” combining the experts’ assessments
Validate the combined assessment:
Evaluate the performance of the weighted combination (“Decision Maker”) using the same scoring as for individual experts
Compare to equal-weight combination and best-performing individual experts
Conduct robustness checks:
Perform cross-validation by using subsets of calibration questions to form weights
Assess how well performance on calibration questions predicts performance on questions of interest
The Classical Model aims to create an optimal aggregate assessment that outperforms both equal-weight combinations and individual experts. By using objective performance measures from calibration questions, it provides a scientifically defensible method for evaluating and synthesizing expert judgment under uncertainty.
Using Data to Support Decisions
ICH Q9(R1) emphasizes the importance of basing risk management decisions on scientific knowledge and data. The guideline encourages organizations to:
Develop robust knowledge management systems to capture and maintain product and process knowledge
Create standardized repositories for technical data and information
Implement systems to collect and convert data into usable knowledge
Gather and analyze relevant data to support risk-based decisions
Use quantitative methods where feasible, such as statistical models or predictive analytics
Specific approaches for using data in QRM may include:
Analyzing historical data on process performance, deviations, and quality issues to inform risk assessments
Employing statistical process control and process capability analysis to evaluate and monitor risks
Utilizing data mining and machine learning techniques to identify patterns and potential risks in large datasets
Implementing real-time data monitoring systems to enable proactive risk management
Conducting formal data quality assessments to ensure decisions are based on reliable information
Digitalization and emerging technologies can support data-driven decision making, but remember that validation requirements for these technologies should not be overlooked.
Improving Risk Assessment Tools
The design of risk assessment tools plays a critical role in minimizing subjectivity. Tools with well-defined scoring criteria and clear guidance on interpreting results can reduce variability in how risks are evaluated. For example, using quantitative methods where feasible—such as statistical models or predictive analytics—can provide more objective insights compared to qualitative scoring systems.
Organizations should also validate their tools periodically to ensure they remain fit-for-purpose and aligned with current regulatory expectations.
Leverage Good Risk Questions
A well-formulated risk question can significantly help reduce subjectivity in quality risk management (QRM) activities. Here’s how a good risk question contributes to reducing subjectivity:
Clarity and Focus
A good risk question provides clarity and focus for the risk assessment process. By clearly defining the scope and context of the risk being evaluated, it helps align all participants on what specifically needs to be assessed. This alignment reduces the potential for individual interpretations and subjective assumptions about the risk scenario.
Specific and Measurable Terms
Effective risk questions use specific and measurable terms rather than vague or ambiguous language. For example, instead of asking “What are the risks to product quality?”, a better question might be “What are the potential causes of out-of-specification dissolution results for Product X in the next 6 months?”. The specificity in the latter question helps anchor the assessment in objective, measurable criteria.
Factual Basis
A well-crafted risk question encourages the use of factual information and data rather than opinions or guesses. It should prompt the risk assessment team to seek out relevant data, historical information, and scientific knowledge to inform their evaluation. This focus on facts and evidence helps minimize the influence of personal biases and subjective judgments.
Standardized Approach
Using a consistent format for risk questions across different assessments promotes a standardized approach to risk identification and analysis. This consistency reduces variability in how risks are framed and evaluated, thereby decreasing the potential for subjective interpretations.
Objective Criteria
Good risk questions often incorporate or imply objective criteria for risk evaluation. For instance, a question like “What factors could lead to a deviation from the acceptable range of 5-10% for impurity Y?” sets clear, objective parameters for the assessment, reducing the room for subjective interpretation of what constitutes a significant risk.
Promotes Structured Thinking
Well-formulated risk questions encourage structured thinking about potential hazards, their causes, and consequences. This structured approach helps assessors focus on objective factors and causal relationships rather than relying on gut feelings or personal opinions.
Facilitates Knowledge Utilization
A good risk question should prompt the assessment team to utilize available knowledge effectively. It encourages the team to draw upon relevant data, past experiences, and scientific understanding, thereby grounding the assessment in objective information rather than subjective impressions.
By crafting risk questions that embody these characteristics, QRM practitioners can significantly reduce the subjectivity in risk assessments, leading to more reliable, consistent, and scientifically sound risk management decisions.
Fostering a Culture of Continuous Improvement
Reducing subjectivity in QRM is an ongoing process that requires a commitment to continuous improvement. Organizations should regularly review their QRM practices to identify areas for enhancement and incorporate feedback from stakeholders. Investing in training programs that build competencies in risk assessment methodologies and decision-making frameworks is essential for sustaining progress.
Moreover, fostering a culture that values transparency, collaboration, and accountability can empower teams to address subjectivity proactively. Encouraging open discussions about uncertainties or disagreements during risk assessments can lead to more robust outcomes.
Conclusion
The revisions introduced in ICH Q9(R1) represent a significant step forward in addressing long-standing challenges associated with subjectivity in QRM. By leveraging knowledge management, implementing structured decision-making processes, addressing cognitive biases, enhancing formality levels appropriately, and improving risk assessment tools, organizations can align their practices with the updated guidelines while ensuring more reliable and science-based outcomes.
It has been two years, it is long past time be be addressing these in your risk management process and quality system.
Ultimately, reducing subjectivity not only strengthens compliance with regulatory expectations but also enhances the quality of pharmaceutical products and safeguards patient safety—a goal that lies at the heart of effective Quality Risk Management.