ICH Q8 (Pharmaceutical Development), Q9 (Quality Risk Management), and Q10 (Pharmaceutical Quality System) provide a comprehensive framework for transforming change management from a reactive compliance exercise into a strategic enabler of quality and innovation.
The ICH Q8-Q10 triad is my favorite framework pharmaceutical quality systems: Q8’s Quality by Design (QbD) principles establish proactive identification of critical quality attributes (CQAs) and design spaces, shifting the paradigm from retrospective testing to prospective control; Q9 provides the scaffolding for risk-based decision-making, enabling organizations to prioritize resources based on severity, occurrence, and detectability of risks; and, Q10 closes the loop by embedding these concepts into a lifecycle-oriented quality system, emphasizing knowledge management and continual improvement.
These guidelines create a robust foundation for change control. Q8 ensures changes align with product and process understanding, Q9 enables risk-informed evaluation, and Q10 mandates systemic integration across the product lifecycle. This triad rejects the notion of change control as a standalone procedure, instead positioning it as a manifestation of organizational quality culture.
The PIC/S Perspective: Risk-Based Change Management
The PIC/S guidance (PI 054-1) reinforces ICH principles by offering a methodology that emphasizes effectiveness as the cornerstone of change management. It outlines four pillars:
- Proposal and Impact Assessment: Systematic evaluation of cross-functional impacts, including regulatory filings, process interdependencies, and stakeholder needs.
- Risk Classification: Stratifying changes as critical/major/minor based on potential effects on product quality, patient safety, and data integrity.
- Implementation with Interim Controls: Bridging current and future states through mitigations like enhanced monitoring or temporary procedural adjustments.
- Effectiveness Verification: Post-implementation reviews using metrics aligned with change objectives, supported by tools like statistical process control (SPC) or continued process verification (CPV).
This guidance operationalizes ICH concepts by mandating traceability from change rationale to verified outcomes, creating accountability loops that prevent “paper compliance.”
A Five-Level Maturity Model for Change Control
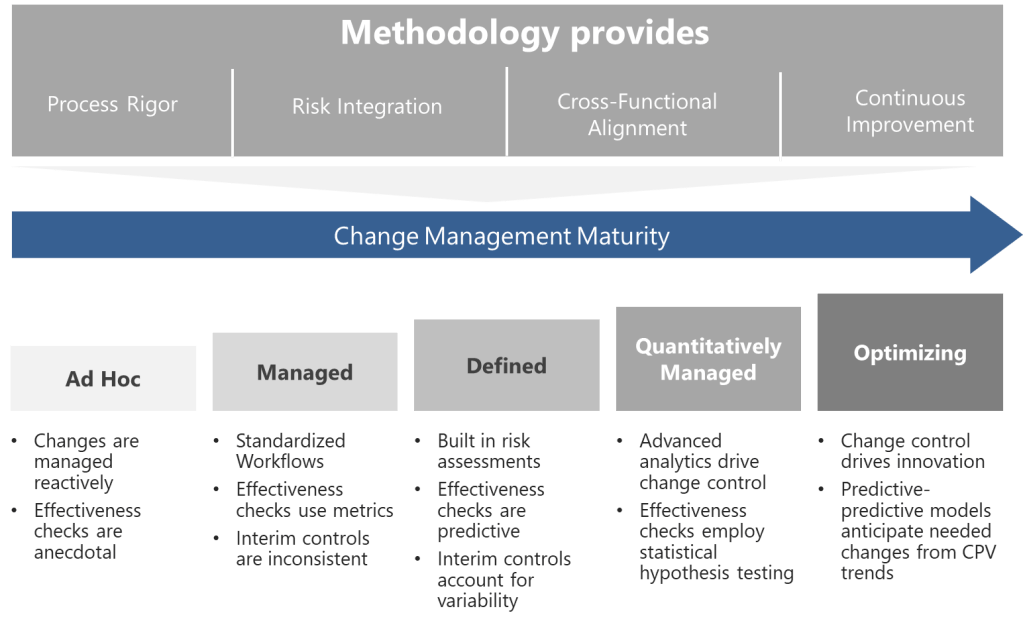
Building on these foundations, I propose a maturity model that evaluates organizational capability across four dimensions, each addressing critical aspects of pharmaceutical change control systems:
- Process Rigor
- Assesses the standardization, documentation, and predictability of change control workflows.
- Higher maturity levels incorporate design space utilization (ICH Q8), automated risk thresholds, and digital tools like Monte Carlo simulations for predictive impact modeling.
- Progresses from ad hoc procedures to AI-driven, self-correcting systems that preemptively identify necessary changes via CPV trends.
- Risk Integration
- Measures how effectively quality risk management (ICH Q9) is embedded into decision-making.
- Includes risk-based classification (critical/major/minor), use of the right tool, and dynamic risk thresholds tied to process capability indices (CpK/PpK).
- At advanced levels, machine learning models predict failure probabilities, enabling proactive mitigations.
- Cross-Functional Alignment
- Evaluates collaboration between QA, regulatory, manufacturing, and supply chain teams during change evaluation.
- Maturity is reflected in centralized review boards, real-time data integration (e.g., ERP/LIMS connectivity), and harmonized procedures across global sites.
- Continuous Improvement
- Tracks the organization’s ability to learn from past changes and innovate.
- Incorporates metrics like “first-time regulatory acceptance rate” and “change-related deviation reduction.”
- Top-tier organizations use post-change data to refine design spaces and update control strategies.
Level 1: Ad Hoc (Chaotic)
At this initial stage, changes are managed reactively. Procedures exist but lack standardization—departments use disparate tools, and decisions rely on individual expertise rather than systematic risk assessment. Effectiveness checks are anecdotal, often reduced to checkbox exercises. Organizations here frequently experience regulatory citations related to undocumented changes or inadequate impact assessments.
Progression Strategy: Begin by mapping all change types and aligning them with ICH Q9 risk principles. Implement a centralized change control procedure with mandatory risk classification.
Level 2: Managed (Departmental)
Changes follow standardized workflows within functions, but silos persist. Risk assessments are performed but lack cross-functional input, leading to unanticipated impacts. Effectiveness checks use basic metrics (e.g., # of changes), yet data analysis remains superficial. Interim controls are applied inconsistently, often overcompensating with excessive conservatism or being their in name only.
Progression Strategy: Establish cross-functional change review boards. Introduce the right level of formality of risk for changes and integrate CPV data into effectiveness reviews.
Level 3: Defined (Integrated)
The organization achieves horizontal integration. Changes trigger automated risk assessments using predefined criteria from ICH Q8 design spaces. Effectiveness checks leverage predictive analytics, comparing post-change performance against historical baselines. Knowledge management systems capture lessons learned, enabling proactive risk identification. Interim controls are fully operational, with clear escalation paths for unexpected variability.
Progression Strategy: Develop a unified change control platform that connects to manufacturing execution systems (MES) and laboratory information management systems (LIMS). Implement real-time dashboards for change-related KPIs.
Level 4: Quantitatively Managed (Predictive)
Advanced analytics drive change control. Machine learning models predict change impacts using historical data, reducing assessment timelines. Risk thresholds dynamically adjust based on process capability indices (CpK/PpK). Effectiveness checks employ statistical hypothesis testing, with sample sizes calculated via power analysis. Regulatory submissions for post-approval changes are partially automated through ICH Q12-enabled platforms.
Progression Strategy: Pilot digital twins for high-complexity changes, simulating outcomes before implementation. Formalize partnerships with regulators for parallel review of major changes.
Level 5: Optimizing (Self-Correcting)
Change control becomes a source of innovation. Predictive-predictive models anticipate needed changes from CPV trends. Change histories provide immutable audit trails across the product. Autonomous effectiveness checks trigger corrective actions via integrated CAPA systems. The organization contributes to industry-wide maturity through participation in various consensus standard and professional associations.
Progression Strategy: Institutionalize a “change excellence” function focused on benchmarking against emerging technologies like AI-driven root cause analysis.
Methodological Pillars: From Framework to Practice
Translating this maturity model into practice requires three methodological pillars:
1. QbD-Driven Change Design
Leverage Q8’s design space concepts to predefine allowable change ranges. Changes outside the design space trigger Q9-based risk assessments, evaluating impacts on CQAs using tools like cause-effect matrices. Fully leverage Q12.
2. Risk-Based Resourcing
Apply Q9’s risk prioritization to allocate resources proportionally. A minor packaging change might require a 2-hour review by QA, while a novel drug product process change engages R&D, regulatory, and supply chain teams in a multi-week analysis. Remember, the “level of effort commensurate with risk” prevents over- or under-management.
3. Closed-Loop Verification
Align effectiveness checks with Q10’s lifecycle approach. Post-change monitoring periods are determined by statistical confidence levels rather than fixed durations. For instance, a formulation change might require 10 consecutive batches within CpK >1.33 before closure. PIC/S-mandated evaluations of unintended consequences are automated through anomaly detection algorithms.
Overcoming Implementation Barriers
Cultural and technical challenges abound in maturity progression. Common pitfalls include:
- Overautomation: Implementing digital tools before standardizing processes, leading to “garbage in, gospel out” scenarios.
- Risk Aversion: Misapplying Q9 to justify excessive controls, stifling continual improvement.
- Siloed Metrics: Tracking change closure rates without assessing long-term quality impacts.
Mitigation strategies involve:
- Co-developing procedures with frontline staff to ensure usability.
- Training on “right-sized” QRM—using ICH Q9 to enable, not hinder, innovation.
- Adopting balanced scorecards that link change metrics to business outcomes (e.g., time-to-market, cost of quality).
The Future State: Change Control as a Competitive Advantage
Change control maturity increasingly differentiates market leaders. Organizations reaching Level 5 capabilities can leverage:
- Adaptive Regulatory Strategies: Real-time submission updates via ICH Q12’s Established Conditions framework.
- AI-Enhanced Decision Making: Predictive analytics for change-related deviations, reducing downstream quality events.
- Patient-Centric Changes: Direct integration of patient-reported outcomes (PROs) into change effectiveness criteria.
Maturity as a Journey, Not a Destination
The proposed model provides a roadmap—not a rigid prescription—for advancing change control. By grounding progression in ICH Q8-Q10 and PIC/S principles, organizations can systematically enhance their change agility while maintaining compliance. Success requires viewing maturity not as a compliance milestone but as a cultural commitment to excellence, where every change becomes an opportunity to strengthen quality and accelerate innovation.
In an era of personalized medicines and decentralized manufacturing, the ability to manage change effectively will separate thriving organizations from those merely surviving. The journey begins with honest self-assessment against this model and a willingness to invest in the systems, skills, and culture that make maturity possible.