Making a Quality Leader: From Theory to Practice
Always a pleasure to hear from the Human Development and Leadership (HDL) technical community. Their competency framework is a great tool that can help many a quality leader as they struggle to build the leadership competencies necessary to strive towards excellence.
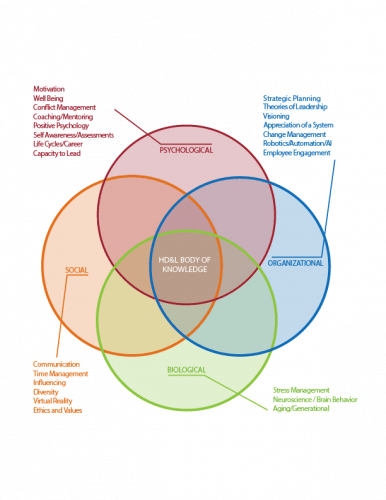
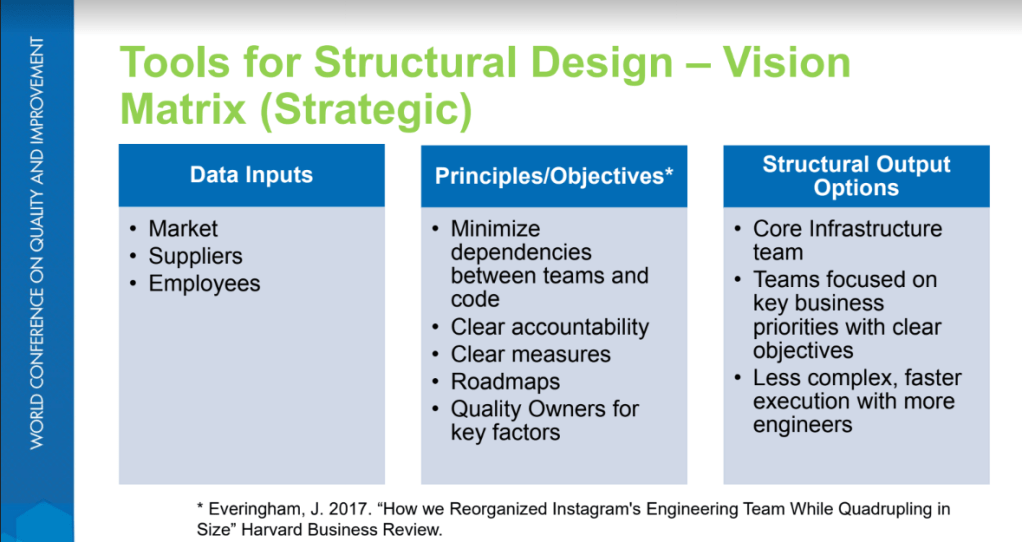
Having done a lot of comptency frameworks I think the HDL has done a good job building theirs out as a progressive framework. I think a challenge is how do we make it easy to scale up to this more detailed HD&LBok from the QBok, and occasionally the same concept can be discussed a few different ways across different technical community boks and this lack of consistency is a detriment.
Stressing practice as the core part of a competency framework. A good conversation at my table was how often these competencies move back a little as we change roles and organizations.
No competency framework is valuable without a good development plan and the workshop did a great job at introducing how to use one.
Honor Deming’s 14 Points Through Modular Kaizen by Grace Duffy
Grace has always been a mentor who always brings thought provoking topics to the society.
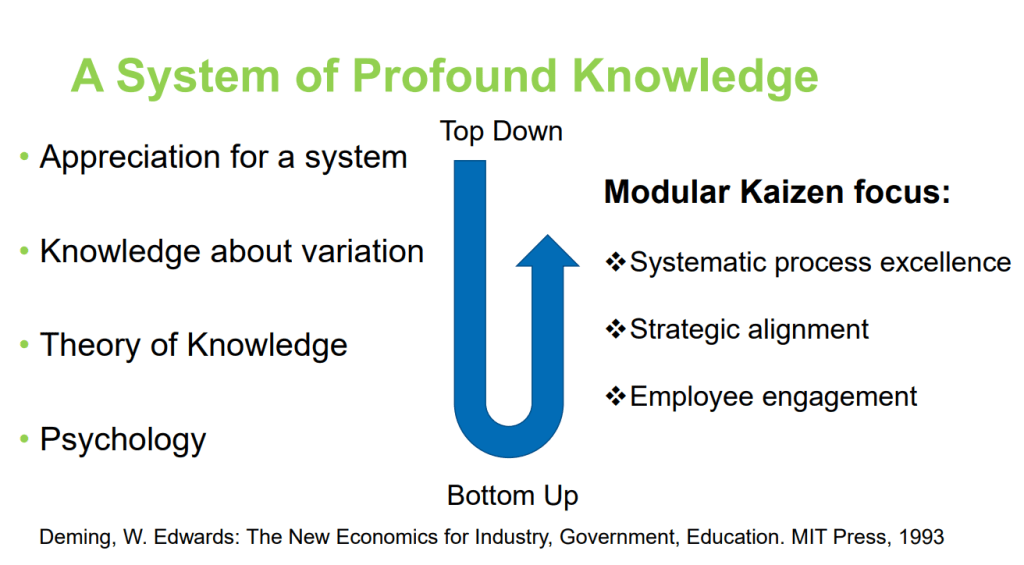
Goes without saying that I’m a fan of the 14 points and the System of Profound Knowledge. I’ve written a lot about Driving out Fear. It ties in a lot of my thoughts about how much of what is happening now in organizational excellence is the evolution from these 14 points.
Looking to the past is an important part of building the future. I think Grace did a great job of respecting the past to drive innovation and new ideas within the quality practice. I loved the care for our past, and the urge to challenge the future in the audience. She really avoided the tendency in some quality circles to obsess about the past (Toyota nuts I am thinking of you) and thus trap the present and stifle the future.
Grace does an amazing job being a pillar of the quality community while still being an iconcolast. When you see her name on the conference session list I always recommend taking the time to attend.
Quality Past & Present
I found most of the videos to be a little personality driven instead of insightful pieces of history. I would hope this session would draw from our history in exciting ways, that was not realized. However the ending charge to create solutions instead of just solving problems is a pivotal one.
2022 Business Meeting
This business meeting drives home the struggle the ASQ has on figuring out what comes next.
As an organization we do not understand what a digital organization looks like. Only 2000 users of the app in the US is embarrassing. Teens can create apps with 100 times more users.
It is nice to admit that actively listening to the members is crucial. I look forward to that consistently happening. We don’t always do a good job of using the tools at the heart of the QBok.
Finances are still rocky, though the Federal Stimulus has left the ASQ in an okay place.
I was actually shocked to hear the chair of the ASQ say it is hard to have metrics for meeting strategic goals. We have entire methodologies dedicated to that.
There are a lot of challenges ahead of us.