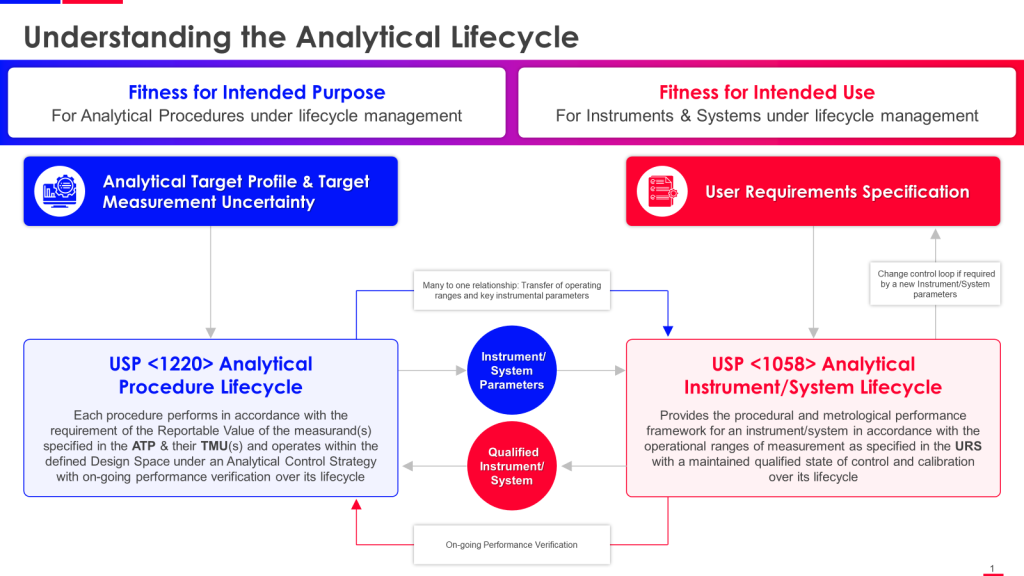
I often get asked why I moved from a broader senior role in Quality Management to a particular but deep role in Quality Engineering and Validation. There are many answers, but the biggest is that validation is poised for some exciting shifts due to navigating a complex validation landscape characterized by rapid technological advancements, evolving regulatory standards, and the development of novel therapies. Addressing these challenges requires innovation, collaboration, and a proactive approach to risk management and data integration. Topics near and dear to me.
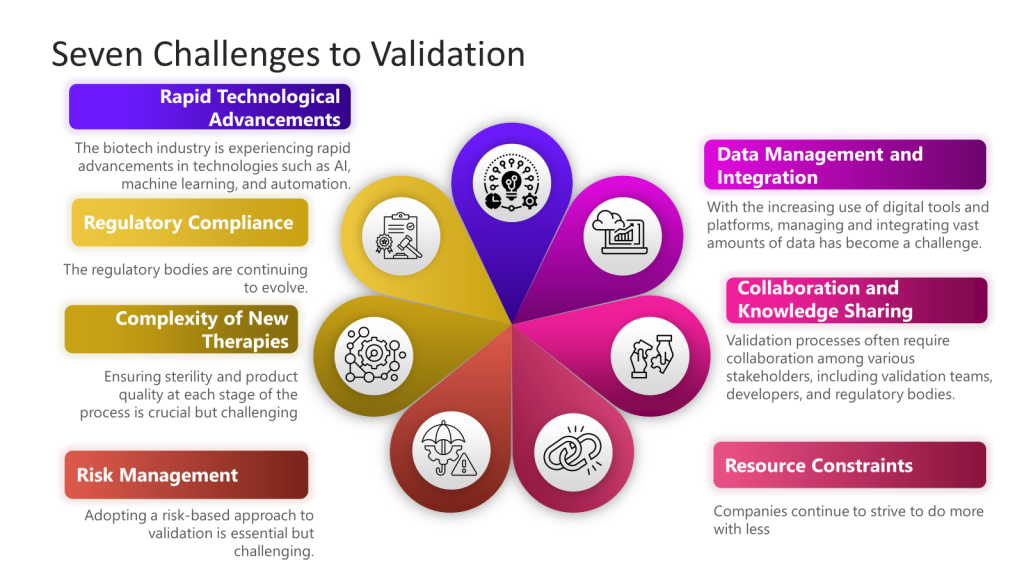
Today’s Challenges in Biotech Validation
1. Rapid Technological Advancements
The biotech industry is experiencing rapid technological advancements such as AI, machine learning, and automation. Integrating these technologies into validation processes can be challenging due to the need for new validation frameworks and methodologies.
2. Regulatory Compliance
Maintaining compliance with evolving regulatory standards is a significant challenge. Regulatory bodies like the FDA continuously update guidelines for technological advancements.
3. Complexity of New Therapies
Developing novel therapies, such as cell and gene therapies, introduces additional complexity to the validation process. These therapies often require redesigned facilities and equipment to accommodate their sensitive and sterile nature. Ensuring sterility and product quality at each process stage is crucial but challenging.
4. Data Management and Integration
Managing and integrating vast amounts of data has become challenging with the increasing use of digital tools and platforms. Effective data management is essential for predictive modeling and risk management in validation processes. Organizations must adopt robust data analytics and machine learning tools to handle this data efficiently.
5. Collaboration and Knowledge Sharing
Validation processes often require collaboration among various stakeholders, including validation teams, developers, and regulatory bodies. Ensuring real-time communication and data sharing can be challenging but is essential for streamlining validation efforts and aligning goals.
6. Resource Constraints
Smaller biotech companies, in particular, face resource constraints regarding funding, personnel, and expertise. These constraints can hinder their ability to implement advanced validation techniques and maintain compliance with regulatory standards.
7. Risk Management
Adopting a risk-based approach to validation is essential but challenging. Companies must identify and mitigate risks throughout the product lifecycle, which requires a thorough understanding of potential risks and effective risk management strategies.
Let’s Avoid the Term Validation 4.0
Let’s avoid the 4.0 term. We are constantly evolving, and adding a current ‘buzziness’ to it does no one any favors. We are shifting from traditional, paper-heavy validation methods to a more dynamic, data-driven, and digitalized process. Yes, we are leveraging modern technologies such as automation, data analytics, artificial intelligence (AI), and the Internet of Things (IoT) to enhance validation processes’ efficiency, flexibility, and reliability. But we don’t need buzziness, we just need to give it some thought, experiment, and refine.