It is rare when a journalist reports on the GMP side of the industry. Most reporting tends to be pretty shallow, and only when a major crisis happens, such as the baby food manufacturing failures. So I love it when a journalist takes the time to understand our field and write a detailed piece. Katherine Eban, who wrote the insightful Bottle of Lies, which I am known to gift copies of, being a great example of a journalist can creditably speak our language and than translate it to the general public.
The article stresses the ongoing crisis in that the FDA does not have enough inspectors, a crisis that keeps getting worse under the current administration.
The Form 483 that is linked should frighten anyone, as it has 3 pages of redacted batches that were shipped to the US.
I share the frustration and concern that Form 483s are not transparent, and that the FDA does not follow the same principle of the average health inspector for local restaurants where I see the grade when I walk in. It is pretty difficult to figure out where a medicine is made, and failing access to some expensive services, can be a real pain to figure out the status of any given manufacturing site.
The Form 483 for Glenmark is, unfortunately, all too common for an Indian generics manufacturing site. Poor validation, no real cleaning, lack of investigations – these are all very serious, and unfortunately recurring.
I am very concerned that the woes of Indian manufacturing sites (and Chinese) will just get worse as the FDA is torn apart by a bunch of authoritarian ideologues who think sunshine and bleach are cure-alls.
Strategic approaches like grouping, family classification, and bracketing are invaluable tools in the validation professional’s toolkit. While these terms are sometimes used interchangeably, they represent distinct strategies with specific applications and regulatory considerations.
Grouping, Family and Bracket
Equipment Grouping – The Broader Approach
Equipment grouping (sometimes called matrixing) represents a broad risk-based approach where multiple equipment items are considered equivalent for validation purposes. This strategy allows companies to optimize validation efforts by categorizing equipment based on design, functionality, and risk profiles. The key principle behind grouping is that equipment with similar characteristics can be validated using a common approach, reducing redundancy in testing and documentation.
Example – Manufacturing
Equipment grouping might apply to multiple buffer preparation tanks that share fundamental design characteristics but differ in volume or specific features. For example, a facility might have six 500L buffer preparation tanks from the same manufacturer, used for various buffer preparations throughout the purification process. These tanks might have identical mixing technologies, materials of construction, and cleaning processes.
Under a grouping approach, the manufacturer could develop one validation plan covering all six tanks. This plan would outline the overall validation strategy, including the rationale for grouping, the specific tests to be performed, and how results will be evaluated across the group. The plan might specify that while all tanks will undergo full Installation Qualification (IQ) to verify proper installation and utility connections, certain Operational Qualification (OQ) and Performance Qualification (PQ) tests can be consolidated.
The mixing efficiency test might be performed on only two tanks (e.g., the first and last installed), with results extrapolated to the entire group. However, critical parameters like temperature control accuracy would still be tested individually for each tank. The grouping approach would also allow for the application of the same cleaning validation protocol across all tanks, with appropriate justification. This might involve developing a worst-case scenario for cleaning validation based on the most challenging buffer compositions and applying the results across all tanks in the group.
Examples – QC
In the QC laboratory setting, equipment grouping might involve multiple identical analytical instruments such as HPLCs used for release testing. For instance, five HPLC systems of the same model, configured with identical detectors and software versions, might be grouped for qualification purposes.
The QC group could justify standardized qualification protocols across all five systems. This would involve developing a comprehensive protocol that covers all aspects of HPLC qualification but allows for efficient execution across the group. For example, software validation might be performed once and applied to all systems, given that they use identical software versions and configurations.
Consolidated performance testing could be implemented where appropriate. This might involve running system suitability tests on a representative sample of HPLCs rather than exhaustively on each system. However, critical performance parameters like detector linearity would still be verified individually for each HPLC to ensure consistency across the group.
Uniform maintenance and calibration schedules could be established for the entire group, simplifying ongoing management and reducing the risk of overlooking maintenance tasks for individual units. This approach ensures consistent performance across all grouped HPLCs while optimizing resource utilization.
Equipment grouping provides broad flexibility but requires careful consideration of which validation elements truly can be shared versus those that must remain equipment-specific. The key to successful grouping lies in thorough risk assessment and scientific justification for any shared validation elements.
Family Approach: Categorizing Based on Common Characteristics
The family approach represents a more structured categorization methodology where equipment is grouped based on specific common characteristics including identical risk classification, common intended purpose, and shared design and manufacturing processes. Family grouping typically applies to equipment from the same manufacturer with minor permissible variations. This approach recognizes that while equipment within a family may not be identical, their core functionalities and critical quality attributes are sufficiently similar to justify a common validation approach with specific considerations for individual variations.
Example – Manufacturing
A family approach might apply to chromatography skids designed for different purification steps but sharing the same basic architecture. For example, three chromatography systems from the same manufacturer might have different column sizes and flow rates but identical control systems, valve technologies, and sensor types.
Under a family approach, base qualification protocols would be identical for all three systems. This core protocol would cover common elements such as control system functionality, alarm systems, and basic operational parameters. Each system would undergo full IQ verification to ensure proper installation, utility connections, and compliance with design specifications. This individual IQ is crucial as it accounts for the specific installation environment and configuration of each unit.
OQ testing would focus on the specific operating parameters for each unit while leveraging a common testing framework. All systems might undergo the same sequence of tests (e.g., flow rate accuracy, pressure control, UV detection linearity), but the acceptance criteria and specific test conditions would be tailored to each system’s operational range. This approach ensures that while the overall qualification strategy is consistent, each system is verified to perform within its specific design parameters.
Shared control system validation could be leveraged across the family. Given that all three systems use identical control software and hardware, a single comprehensive software validation could be performed and applied to all units. This might include validation of user access controls, data integrity features, and critical control algorithms. However, system-specific configuration settings would still need to be verified individually.
Example – QC
In QC testing, a family approach could apply to dissolution testers that serve the same fundamental purpose but have different configurations. For instance, four dissolution testers might have the same underlying technology and control systems but different numbers of vessels or sampling configurations.
The qualification strategy could include common template protocols with configuration-specific appendices. This approach allows for a standardized core qualification process while accommodating the unique features of each unit. The core protocol might cover elements common to all units, such as temperature control accuracy, stirring speed precision, and basic software functionality.
Full mechanical verification would be performed for each unit to account for the specific configuration of vessels and sampling systems. This ensures that despite being part of the same family, each unit’s unique physical setup is thoroughly qualified.
A shared software validation approach could be implemented, focusing on the common control software used across all units. This might involve validating core software functions, data processing algorithms, and report generation features. However, configuration-specific software settings and any unique features would require individual verification.
Configuration-specific performance testing would be conducted to address the unique aspects of each unit. For example, a dissolution tester with automated sampling would undergo additional qualification of its sampling system, while units with different numbers of vessels might require specific testing to ensure uniform performance across all vessels.
The family approach provides a middle ground, recognizing fundamental similarities while still acknowledging equipment-specific variations that must be qualified independently. This strategy is particularly useful in biologics manufacturing and QC, where equipment often shares core technologies but may have variations to accommodate different product types or analytical methods.
Bracketing Approach: Strategic Testing Reduction
Bracketing represents the most targeted approach, involving the selective testing of representative examples from a group of identical equipment to reduce the overall validation burden. This approach requires rigorous scientific justification and risk assessment to demonstrate that the selected “brackets” truly represent the performance of all units. Bracketing is based on the principle that if the extreme cases (brackets) meet acceptance criteria, units falling within these extremes can be assumed to comply as well.
Example – Manufacturing
Bracketing might apply to multiple identical bioreactors. For example, a facility might have six 2000L single-use bioreactors of identical design, from the same manufacturing lot, installed in similar environments, and operated by the same control system.
Under a bracketing approach, all bioreactors would undergo basic installation verification to ensure proper setup and connection to utilities. This step is crucial to confirm that each unit is correctly installed and ready for operation, regardless of its inclusion in comprehensive testing.
Only two bioreactors (typically the minimum and maximum in the installation sequence) might undergo comprehensive OQ testing. This could include detailed evaluation of temperature control systems, agitation performance, gas flow accuracy, and pH/DO sensor functionality. The justification for this approach would be based on the identical design and manufacturing of the units, with the assumption that any variation due to manufacturing or installation would be most likely to manifest in the first or last installed unit.
Temperature mapping might be performed on only two units with justification that these represent “worst-case” positions. For instance, the units closest to and farthest from the HVAC outlets might be selected for comprehensive temperature mapping studies. These studies would involve placing multiple temperature probes throughout the bioreactor vessel and running temperature cycles to verify uniform temperature distribution and control.
Process performance qualification might be performed on a subset of reactors. This could involve running actual production processes (or close simulations) on perhaps three of the six reactors – for example, the first installed, the middle unit, and the last installed. These runs would evaluate critical process parameters and quality attributes to demonstrate consistent performance across the bracketed group.
Example – QC
Bracketing might apply to a set of identical incubators used for microbial testing. For example, eight identical incubators might be installed in the same laboratory environment.
The bracketing strategy could include full IQ documentation for all units to ensure proper installation and basic functionality. This step verifies that each incubator is correctly set up, connected to appropriate utilities, and passes basic operational checks.
Comprehensive temperature mapping would be performed for only the first and last installed units. This intensive study would involve placing calibrated temperature probes throughout the incubator chamber and running various temperature cycles to verify uniform heat distribution and precise temperature control. The selection of the first and last units is based on the assumption that any variations due to manufacturing or installation would be most likely to appear in these extreme cases.
Challenge testing on a subset representing different locations in the laboratory might be conducted. This could involve selecting incubators from different areas of the lab (e.g., near windows, doors, or HVAC vents) for more rigorous performance testing. These tests might include recovery time studies after door openings, evaluation of temperature stability under various load conditions, and assessment of humidity control (if applicable).
Ongoing monitoring that continuously verifies the validity of the bracketing approach would be implemented. This might involve rotating additional performance tests among all units over time or implementing a program of periodic reassessment to confirm that the bracketed approach remains valid. For instance, annual temperature distribution studies might be rotated among all incubators, with any significant deviations triggering a reevaluation of the bracketing strategy.
Key Differences and Selection Criteria
The primary differences between these approaches can be summarized in several key areas:
Scope and Application
Grouping is the broadest approach, applicable to equipment with similar functionality but potential design variations. This strategy is most useful when dealing with a wide range of equipment that serves similar purposes but may have different manufacturers or specific features. For example, in a large biologics facility, grouping might be applied to various types of pumps used throughout the manufacturing process. While these pumps may have different flow rates or pressure capabilities, they could be grouped based on their common function of fluid transfer and similar cleaning requirements.
The Family approach is an intermediate strategy, applicable to equipment with common design principles and minor variations. This is particularly useful for equipment from the same manufacturer or product line, where core technologies are shared but specific configurations may differ. In a QC laboratory, a family approach might be applied to a range of spectrophotometers from the same manufacturer. These instruments might share the same fundamental optical design and software platform but differ in features like sample capacity or specific wavelength ranges.
Bracketing is the most focused approach, applicable only to identical equipment with strong scientific justification. This strategy is best suited for situations where multiple units of the exact same equipment model are installed under similar conditions. For example, in a fill-finish operation, bracketing might be applied to a set of identical lyophilizers installed in the same clean room environment.
Testing Requirements
In a Grouping approach, each piece typically requires individual testing, but with standardized protocols. This means that while the overall validation strategy is consistent across the group, specific tests are still performed on each unit to account for potential variations. For instance, in a group of buffer preparation tanks, each tank would undergo individual testing for critical parameters like temperature control and mixing efficiency, but using a standardized testing protocol developed for the entire group.
The Family approach involves core testing that is standardized, with variations to address equipment-specific features. This allows for a more efficient validation process where common elements are tested uniformly across the family, while specific features of each unit are addressed separately. In the case of a family of chromatography systems, core functions like pump operation and detector performance might be tested using identical protocols, while specific column compatibility or specialized detection modes would be validated individually for units that possess these features.
Bracketing involves selective testing of representative units with extrapolation to the remaining units. This approach significantly reduces the overall testing burden but requires robust justification. For example, in a set of identical bioreactors, comprehensive performance testing might be conducted on only the first and last installed units, with results extrapolated to the units in between. However, this approach necessitates ongoing monitoring to ensure the continued validity of the extrapolation.
Documentation Needs
Grouping requires individual documentation with cross-referencing to shared elements. Each piece of equipment within the group would have its own validation report, but these reports would reference a common validation master plan and shared testing protocols. This approach ensures that while each unit is individually accounted for, the efficiency gains of the grouping strategy are reflected in the documentation.
The Family approach typically involves standardized core documentation with equipment-specific supplements. This might manifest as a master validation report for the entire family, with appendices or addenda addressing the specific features or configurations of individual units. This structure allows for efficient document management while still providing a complete record for each piece of equipment.
Bracketing necessitates a comprehensive justification document plus detailed documentation for tested units. This approach requires the most rigorous upfront documentation to justify the bracketing strategy, including risk assessments and scientific rationale. The validation reports for the tested “bracket” units would be extremely detailed, as they serve as the basis for qualifying the entire set of equipment.
Risk Assessment
In a Grouping approach, the risk assessment is focused on demonstrating equivalence for specific validation purposes. This involves a detailed analysis of how variations within the group might impact critical quality attributes or process parameters. The risk assessment must justify why certain tests can be standardized across the group and identify any equipment-specific risks that need individual attention.
For the Family approach, risk assessment is centered on evaluating permissible variations within the family. This involves a thorough analysis of how differences in specific features or configurations might impact equipment performance or product quality. The risk assessment must clearly delineate which aspects of validation can be shared across the family and which require individual consideration.
Bracketing requires the most rigorous risk assessment to justify the extrapolation of results from tested units to non-tested units. This involves a comprehensive evaluation of potential sources of variation between units, including manufacturing tolerances, installation conditions, and operational factors. The risk assessment must provide a strong scientific basis
Criteria
Group Approach
Family Approach
Bracket Approach
Scope and Application
Broadest approach. Applicable to equipment with similar functionality but potential design variations.
Intermediate approach. Applicable to equipment with common design principles and minor variations.
Most focused approach. Applicable only to identical equipment with strong scientific justification.
Equipment Similarity
Similar functionality, potentially different manufacturers or features.
Same manufacturer or product line, core technologies shared, specific configurations may differ.
Identical equipment models installed under similar conditions.
Testing Requirements
Each piece requires individual testing, but with standardized protocols.
Core testing is standardized, with variations to address equipment-specific features.
Selective testing of representative units with extrapolation to the remaining units.
Documentation Needs
Individual documentation with cross-referencing to shared elements.
Standardized core documentation with equipment-specific supplements.
Comprehensive justification document plus detailed documentation for tested units.
Risk Assessment Focus
Demonstrating equivalence for specific validation purposes.
Evaluating permissible variations within the family.
Most rigorous assessment to justify extrapolation of results.
Flexibility
High flexibility to accommodate various equipment types.
Moderate flexibility within a defined family of equipment.
Low flexibility, requires high degree of equipment similarity.
Resource Efficiency
Moderate efficiency gains through standardized protocols.
High efficiency for core validation elements, with specific testing as needed.
Highest potential for efficiency, but requires strong justification.
Regulatory Considerations
Generally accepted with proper justification.
Well-established approach, often preferred for equipment from same manufacturer.
Requires most robust scientific rationale and ongoing verification.
Ideal Use Case
Large facilities with diverse equipment serving similar functions.
Product lines with common core technology but varying features.
Multiple identical units in same facility or laboratory.
Review and document updates as needed (including reapprovals)
Managing changes and revision status
Ensuring availability of current versions
Maintaining document legibility and identification
Controlling distribution of external documents
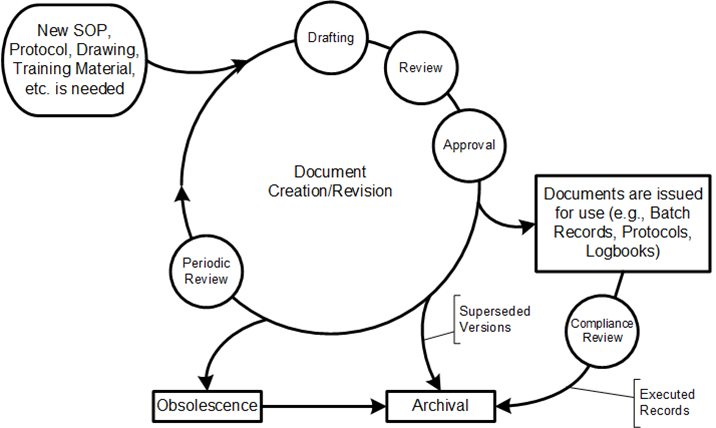
This lifecycle usually has three critical dates associated with approval:
Approval Date: When designated authorities have reviewed and approved the document
Issuance Date: When the document is released into the document management system
Effective Date: When the document officially takes effect and must be followed
These dates are dependent on the type of document and can change as a result of workflow decisions.
Type of Document
Approval Date
Issuance date
Effective Date
Functional
Date Approved by final approver (sequential or parallel)
Date Training Made Available
End of Training Period
Record
Date Approved by final approver (sequential or parallel)
Usually automated to be same as Date Approved
Usually same as Date Approved
Report
Date Approved by final approver (sequential or parallel)
Usually automated to be same as Date Approved
Usually same as Date Approved
At the heart of the difference between these three days is the question of implementation and the Effective Date. At its core, the effective date is the date on which the requirements, instructions, or obligations in a document become binding for all affected parties. In the context of GxP document management, this represents the moment when:
Previous versions of the document are officially superseded
All operations must follow the new procedures outlined in the document
Training on the new procedures must be completed
Compliance audits will use the new document as their reference standard
One of the most frequently overlooked aspects of document management is the implementation period between document approval and its effective date. This period serves a critical purpose: ensuring that all affected personnel understand the document’s content and can execute its requirements correctly before it becomes binding.
In order to implement a new process change in a compliant manner, people must be trained in the new procedure before the document becomes effective. This fundamental principle ensures that by the time a new process goes “live,” everyone is prepared to perform the revised activity correctly and training records have been completed. Without this preparation period, organizations risk introducing non-compliance at the very moment they attempt to improve quality.
The implementation period bridges the gap between formal approval and practical application, addressing the human element of quality systems that automated solutions alone cannot solve.
Selecting Appropriate Implementation Periods
When configuring document change control systems, organizations must establish clear guidelines for determining implementation periods. The most effective approach is to build this determination into the change control workflow itself.
Several factors should influence the selection of implementation periods:
Urgency: In cases of immediate risk to patient safety or product quality, implementation periods may be compressed while still ensuring adequate training.
Risk Assessment: Higher-risk changes typically require more extensive training and therefore longer implementation periods.
Operational Impact: Changes affecting critical operations may need carefully staged implementation.
Training Complexity: Documents requiring hands-on training necessitate longer periods than read-only procedures.
Resource Availability: Consider the availability of trainers and affected personnel
Determining Appropriate Training Periods
The time required for training should be determined during the impact assessment phase of the change approval process. This assessment should consider:
The number of people requiring training
The complexity of the procedural changes
The type of training required (read-only versus observed assessment)
Operational constraints (shift patterns, production schedules)
Many organizations standardize on a default period (typically two weeks), but the most effective approach tailors the implementation period to each document’s specific requirements. For critical processes with many stakeholders, longer periods may be necessary, while simple updates affecting few staff might require only minimal time.
Consider this scenario: Your facility operates two shifts with 70 people during the day and 30 at night. An updated SOP requires all operators to complete not just read-only training but also a one-hour classroom assessment. If manufacturing schedules permit only 10 operators per shift to attend training, you would need a minimum of 7 days before the document becomes effective. Without this calculated implementation period, every operator would instantly become non-compliant when the new procedure takes effect.
The distinction between a procedure’s approval date and its effective date serves a critical purpose. This gap allows for proper training and implementation before the procedure becomes binding. However, there are specific circumstances when personnel might appropriately use a procedure they’ve been trained on before its official effective date.
1. Urgent Safety or Quality Concerns
When there is an immediate risk to patient safety or product quality, the time between approval and effectiveness may be compressed. For these cases there should be a mechanism to move up the effective date.
In such cases, the organization should prioritize training and implementation while still maintaining proper documentation of the accelerated timeline.
2. During Implementation Period for Training Purposes
The implementation period itself is designed to allow for training and controlled introduction of the new procedure. During this time, a limited number of trained personnel may need to use the new procedure to:
Train others on the new requirements
Test the procedure in a controlled environment
Prepare systems and equipment for the full implementation
These are all tasks that should be captured in the change control.
3. For Qualification and Validation Activities
During qualification protocol execution, procedures that have been approved but are not yet effective may be used under controlled conditions to validate systems, equipment, or processes. These activities typically occur before full implementation and are carefully documented to demonstrate compliance. Again these are captured in the change control and appropriate validation plan.
In some regulatory contexts, such as IRB approvals in clinical research, there are provisions for “approval with conditions” where certain activities may proceed before all requirements are finalized2. While not directly analogous to procedure implementation, this demonstrates regulatory recognition of staged implementation approaches.
Required Controls When Using Pre-Effective Procedures
If an organization determines it necessary to use an approved but not yet effective procedure, the following controls should be in place:
Documented Risk Assessment: A risk assessment should be conducted and documented to justify the early use of the procedure, especially considering potential impacts on product quality, data integrity, or patient safety.
Authorization: Special authorization from management and quality assurance should be obtained and documented.
Verification of Training: Evidence must be available confirming that the individuals using the procedure have been properly trained and assessed on the new requirements.
What About Parallel Compliance with Current Effective Procedures?
In all cases, the currently effective procedure must still be followed until the new procedure’s effective date. However there are changes, usually as a result of process improvement, usually in knowledge work processes where it is possible to use parts of the new procedure. For example, the new version of the deviation procedure adds additional requirements for assessing the deviation, or a new risk management tool is rolled out. In these cases you can meet the new compliance path without violating the current compliance path. The organization should demonstrate how both compliance paths are being maintained.
In cases where the new compliance path does not contain the old, but instead offers a new pathway, it is critical to maintain one way of work-as-prescribed and the effective date is a solid line.
Organizations should remember that the implementation period exists to ensure a smooth, compliant transition between procedures. Any exception to this standard approach should be carefully considered, well-justified, and thoroughly documented to maintain GxP compliance and minimize regulatory risk.
We live in a fascinating inflection point in quality management, caught between traditional document-centric approaches and the emerging imperative for data-centricity needed to fully realize the potential of digital transformation. For several decades, we’ve been in a process that continues to accelerate through a technology transition that will deliver dramatic improvements in operations and quality. This transformation is driven by three interconnected trends: Pharma 4.0, the Rise of AI, and the shift from Documents to Data.
The History and Evolution of Documents in Quality Management
The history of document management can be traced back to the introduction of the file cabinet in the late 1800s, providing a structured way to organize paper records. Quality management systems have even deeper roots, extending back to medieval Europe when craftsman guilds developed strict guidelines for product inspection. These early approaches established the document as the fundamental unit of quality management—a paradigm that persisted through industrialization and into the modern era.
The document landscape took a dramatic turn in the 1980s with the increasing availability of computer technology. The development of servers allowed organizations to store documents electronically in centralized mainframes, marking the beginning of electronic document management systems (eDMS). Meanwhile, scanners enabled conversion of paper documents to digital format, and the rise of personal computers gave businesses the ability to create and store documents directly in digital form.
In traditional quality systems, documents serve as the backbone of quality operations and fall into three primary categories: functional documents (providing instructions), records (providing evidence), and reports (providing specific information). This document trinity has established our fundamental conception of what a quality system is and how it operates—a conception deeply influenced by the physical limitations of paper.
Breaking the Paper Paradigm: Limitations of Document-Centric Thinking
The Paper-on-Glass Dilemma
The maturation path for quality systems typically progresses mainly from paper execution to paper-on-glass to end-to-end integration and execution. However, most life sciences organizations remain stuck in the paper-on-glass phase of their digital evolution. They still rely on the paper-on-glass data capture method, where digital records are generated that closely resemble the structure and layout of a paper-based workflow. In general, the wider industry is still reluctant to transition away from paper-like records out of process familiarity and uncertainty of regulatory scrutiny.
Paper-on-glass systems present several specific limitations that hamper digital transformation:
Constrained design flexibility: Data capture is limited by the digital record’s design, which often mimics previous paper formats rather than leveraging digital capabilities. A pharmaceutical batch record system that meticulously replicates its paper predecessor inherently limits the system’s ability to analyze data across batches or integrate with other quality processes.
Manual data extraction requirements: When data is trapped in digital documents structured like paper forms, it remains difficult to extract. This means data from paper-on-glass records typically requires manual intervention, substantially reducing data utilization effectiveness.
Elevated error rates: Many paper-on-glass implementations lack sufficient logic and controls to prevent avoidable data capture errors that would be eliminated in truly digital systems. Without data validation rules built into the capture process, quality systems continue to allow errors that must be caught through manual review.
Unnecessary artifacts: These approaches generate records with inflated sizes and unnecessary elements, such as cover pages that serve no functional purpose in a digital environment but persist because they were needed in paper systems.
Cumbersome validation: Content must be fully controlled and managed manually, with none of the advantages gained from data-centric validation approaches.
Broader Digital Transformation Struggles
Pharmaceutical and medical device companies must navigate complex regulatory requirements while implementing new digital systems, leading to stalling initiatives. Regulatory agencies have historically relied on document-based submissions and evidence, reinforcing document-centric mindsets even as technology evolves.
Beyond Paper-on-Glass: What Comes Next?
What comes after paper-on-glass? The natural evolution leads to end-to-end integration and execution systems that transcend document limitations and focus on data as the primary asset. This evolution isn’t merely about eliminating paper—it’s about reconceptualizing how we think about the information that drives quality management.
In fully integrated execution systems, functional documents and records become unified. Instead of having separate systems for managing SOPs and for capturing execution data, these systems bring process definitions and execution together. This approach drives up reliability and drives out error, but requires fundamentally different thinking about how we structure information.
A prime example of moving beyond paper-on-glass can be seen in advanced Manufacturing Execution Systems (MES) for pharmaceutical production. Rather than simply digitizing batch records, modern MES platforms incorporate AI, IIoT, and Pharma 4.0 principles to provide the right data, at the right time, to the right team. These systems deliver meaningful and actionable information, moving from merely connecting devices to optimizing manufacturing and quality processes.
AI-Powered Documentation: Breaking Through with Intelligent Systems
A dramatic example of breaking free from document constraints comes from Novo Nordisk’s use of AI to draft clinical study reports. The company has taken a leap forward in pharmaceutical documentation, putting AI to work where human writers once toiled for weeks. The Danish pharmaceutical company is using Claude, an AI model by Anthropic, to draft clinical study reports—documents that can stretch hundreds of pages.
This represents a fundamental shift in how we think about documents. Rather than having humans arrange data into documents manually, we can now use AI to generate high-quality documents directly from structured data sources. The document becomes an output—a view of the underlying data—rather than the primary artifact of the quality system.
Data Requirements: The Foundation of Modern Quality Systems in Life Sciences
Shifting from document-centric to data-centric thinking requires understanding that documents are merely vessels for data—and it’s the data that delivers value. When we focus on data requirements instead of document types, we unlock new possibilities for quality management in regulated environments.
At its core, any quality process is a way to realize a set of requirements. These requirements come from external sources (regulations, standards) and internal needs (efficiency, business objectives). Meeting these requirements involves integrating people, procedures, principles, and technology. By focusing on the underlying data requirements rather than the documents that traditionally housed them, life sciences organizations can create more flexible, responsive quality systems.
ICH Q9(R1) emphasizes that knowledge is fundamental to effective risk management, stating that “QRM is part of building knowledge and understanding risk scenarios, so that appropriate risk control can be decided upon for use during the commercial manufacturing phase.” We need to recognize the inverse relationship between knowledge and uncertainty in risk assessment. As ICH Q9(R1) notes, uncertainty may be reduced “via effective knowledge management, which enables accumulated and new information (both internal and external) to be used to support risk-based decisions throughout the product lifecycle.”
This approach helps us ensure that our tools take into account that our processes are living and breathing, our tools should take that into account. This is all about moving to a process repository and away from a document mindset.
Documents as Data Views: Transforming Quality System Architecture
When we shift our paradigm to view documents as outputs of data rather than primary artifacts, we fundamentally transform how quality systems operate. This perspective enables a more dynamic, interconnected approach to quality management that transcends the limitations of traditional document-centric systems.
Breaking the Document-Data Paradigm
Traditionally, life sciences organizations have thought of documents as containers that hold data. This subtle but profound perspective has shaped how we design quality systems, leading to siloed applications and fragmented information. When we invert this relationship—seeing data as the foundation and documents as configurable views of that data—we unlock powerful capabilities that better serve the needs of modern life sciences organizations.
The Benefits of Data-First, Document-Second Architecture
When documents become outputs—dynamic views of underlying data—rather than the primary focus of quality systems, several transformative benefits emerge.
First, data becomes reusable across multiple contexts. The same underlying data can generate different documents for different audiences or purposes without duplication or inconsistency. For example, clinical trial data might generate regulatory submission documents, internal analysis reports, and patient communications—all from a single source of truth.
Second, changes to data automatically propagate to all relevant documents. In a document-first system, updating information requires manually changing each affected document, creating opportunities for errors and inconsistencies. In a data-first system, updating the central data repository automatically refreshes all document views, ensuring consistency across the quality ecosystem.
Third, this approach enables more sophisticated analytics and insights. When data exists independently of documents, it can be more easily aggregated, analyzed, and visualized across processes.
In this architecture, quality management systems must be designed with robust data models at their core, with document generation capabilities built on top. This might include:
A unified data layer that captures all quality-related information
Flexible document templates that can be populated with data from this layer
Dynamic relationships between data entities that reflect real-world connections between quality processes
Powerful query capabilities that enable users to create custom views of data based on specific needs
The resulting system treats documents as what they truly are: snapshots of data formatted for human consumption at specific moments in time, rather than the authoritative system of record.
Electronic Quality Management Systems (eQMS): Beyond Paper-on-Glass
Electronic Quality Management Systems have been adopted widely across life sciences, but many implementations fail to realize their full potential due to document-centric thinking. When implementing an eQMS, organizations often attempt to replicate their existing document-based processes in digital form rather than reconceptualizing their approach around data.
Current Limitations of eQMS Implementations
Document-centric eQMS systems treat functional documents as discrete objects, much as they were conceived decades ago. They still think it terms of SOPs being discrete documents. They structure workflows, such as non-conformances, CAPAs, change controls, and design controls, with artificial gaps between these interconnected processes. When a manufacturing non-conformance impacts a design control, which then requires a change control, the connections between these events often remain manual and error-prone.
This approach leads to compartmentalized technology solutions. Organizations believe they can solve quality challenges through single applications: an eQMS will solve problems in quality events, a LIMS for the lab, an MES for manufacturing. These isolated systems may digitize documents but fail to integrate the underlying data.
Data-Centric eQMS Approaches
We are in the process of reimagining eQMS as data platforms rather than document repositories. A data-centric eQMS connects quality events, training records, change controls, and other quality processes through a unified data model. This approach enables more effective risk management, root cause analysis, and continuous improvement.
For instance, when a deviation is recorded in a data-centric system, it automatically connects to relevant product specifications, equipment records, training data, and previous similar events. This comprehensive view enables more effective investigation and corrective action than reviewing isolated documents.
Looking ahead, AI-powered eQMS solutions will increasingly incorporate predictive analytics to identify potential quality issues before they occur. By analyzing patterns in historical quality data, these systems can alert quality teams to emerging risks and recommend preventive actions.
Manufacturing Execution Systems (MES): Breaking Down Production Data Silos
Manufacturing Execution Systems face similar challenges in breaking away from document-centric paradigms. Common MES implementation challenges highlight the limitations of traditional approaches and the potential benefits of data-centric thinking.
MES in the Pharmaceutical Industry
Manufacturing Execution Systems (MES) aggregate a number of the technologies deployed at the MOM level. MES as a technology has been successfully deployed within the pharmaceutical industry and the technology associated with MES has matured positively and is fast becoming a recognized best practice across all life science regulated industries. This is borne out by the fact that green-field manufacturing sites are starting with an MES in place—paperless manufacturing from day one.
The amount of IT applied to an MES project is dependent on business needs. At a minimum, an MES should strive to replace paper batch records with an Electronic Batch Record (EBR). Other functionality that can be applied includes automated material weighing and dispensing, and integration to ERP systems; therefore, helping the optimization of inventory levels and production planning.
Beyond Paper-on-Glass in Manufacturing
In pharmaceutical manufacturing, paper batch records have traditionally documented each step of the production process. Early electronic batch record systems simply digitized these paper forms, creating “paper-on-glass” implementations that failed to leverage the full potential of digital technology.
Advanced Manufacturing Execution Systems are moving beyond this limitation by focusing on data rather than documents. Rather than digitizing batch records, these systems capture manufacturing data directly, using sensors, automated equipment, and operator inputs. This approach enables real-time monitoring, statistical process control, and predictive quality management.
An example of a modern MES solution fully compliant with Pharma 4.0 principles is the Tempo platform developed by Apprentice. It is a complete manufacturing system designed for life sciences companies that leverages cloud technology to provide real-time visibility and control over production processes. The platform combines MES, EBR, LES (Laboratory Execution System), and AR (Augmented Reality) capabilities to create a comprehensive solution that supports complex manufacturing workflows.
Electronic Validation Management Systems (eVMS): Transforming Validation Practices
Validation represents a critical intersection of quality management and compliance in life sciences. The transition from document-centric to data-centric approaches is particularly challenging—and potentially rewarding—in this domain.
Current Validation Challenges
Traditional validation approaches face several limitations that highlight the problems with document-centric thinking:
Integration Issues: Many Digital Validation Tools (DVTs) remain isolated from Enterprise Document Management Systems (eDMS). The eDMS system is typically the first step where vendor engineering data is imported into a client system. However, this data is rarely validated once—typically departments repeat this validation step multiple times, creating unnecessary duplication.
Validation for AI Systems: Traditional validation approaches are inadequate for AI-enabled systems. Traditional validation processes are geared towards demonstrating that products and processes will always achieve expected results. However, in the digital “intellectual” eQMS world, organizations will, at some point, experience the unexpected.
Continuous Compliance: A significant challenge is remaining in compliance continuously during any digital eQMS-initiated change because digital systems can update frequently and quickly. This rapid pace of change conflicts with traditional validation approaches that assume relative stability in systems once validated.
Data-Centric Validation Solutions
Modern electronic Validation Management Systems (eVMS) solutions exemplify the shift toward data-centric validation management. These platforms introduce AI capabilities that provide intelligent insights across validation activities to unlock unprecedented operational efficiency. Their risk-based approach promotes critical thinking, automates assurance activities, and fosters deeper regulatory alignment.
We need to strive to leverage the digitization and automation of pharmaceutical manufacturing to link real-time data with both the quality risk management system and control strategies. This connection enables continuous visibility into whether processes are in a state of control.
The 11 Axes of Quality 4.0
LNS Research has identified 11 key components or “axes” of the Quality 4.0 framework that organizations must understand to successfully implement modern quality management:
Data: In the quality sphere, data has always been vital for improvement. However, most organizations still face lags in data collection, analysis, and decision-making processes. Quality 4.0 focuses on rapid, structured collection of data from various sources to enable informed and agile decision-making.
Analytics: Traditional quality metrics are primarily descriptive. Quality 4.0 enhances these with predictive and prescriptive analytics that can anticipate quality issues before they occur and recommend optimal actions.
Connectivity: Quality 4.0 emphasizes the connection between operating technology (OT) used in manufacturing environments and information technology (IT) systems including ERP, eQMS, and PLM. This connectivity enables real-time feedback loops that enhance quality processes.
Collaboration: Breaking down silos between departments is essential for Quality 4.0. This requires not just technological integration but cultural changes that foster teamwork and shared quality ownership.
App Development: Quality 4.0 leverages modern application development approaches, including cloud platforms, microservices, and low/no-code solutions to rapidly deploy and update quality applications.
Scalability: Modern quality systems must scale efficiently across global operations while maintaining consistency and compliance.
Management Systems: Quality 4.0 integrates with broader management systems to ensure quality is embedded throughout the organization.
Compliance: While traditional quality focused on meeting minimum requirements, Quality 4.0 takes a risk-based approach to compliance that is more proactive and efficient.
Culture: Quality 4.0 requires a cultural shift that embraces digital transformation, continuous improvement, and data-driven decision-making.
Leadership: Executive support and vision are critical for successful Quality 4.0 implementation.
Competency: New skills and capabilities are needed for Quality 4.0, requiring significant investment in training and workforce development.
The Future of Quality Management in Life Sciences
The evolution from document-centric to data-centric quality management represents a fundamental shift in how life sciences organizations approach quality. While documents will continue to play a role, their purpose and primacy are changing in an increasingly data-driven world.
By focusing on data requirements rather than document types, organizations can build more flexible, responsive, and effective quality systems that truly deliver on the promise of digital transformation. This approach enables life sciences companies to maintain compliance while improving efficiency, enhancing product quality, and ultimately delivering better outcomes for patients.
The journey from documents to data is not merely a technical transition but a strategic evolution that will define quality management for decades to come. As AI, machine learning, and process automation converge with quality management, the organizations that successfully embrace data-centricity will gain significant competitive advantages through improved agility, deeper insights, and more effective compliance in an increasingly complex regulatory landscape.
The paper may go, but the document—reimagined as structured data that enables insight and action—will continue to serve as the foundation of effective quality management. The key is recognizing that documents are vessels for data, and it’s the data that drives value in the organization.
As an American Pharmaceutical Quality professional who has worked in and with European colleagues for decades, I am used to hearing, “But the requirements in country X are different,” to which my response is always, “Prove it.”
EudraLex represents the cornerstone of Good Manufacturing Practice (GMP) regulations within the European Union, providing a comprehensive framework that ensures medicinal products meet stringent quality, safety, and efficacy standards. You will understand the fundamentals if you know and understand Eudralex volume 4. However, despite this unified approach, a few specific national differences exist in how a select few of these regulations are interpreted and implemented – mostly around Qualified Persons, GMP certifications, registrations and inspection types.
EudraLex: The European Union Pharmaceutical Regulatory Framework
EudraLex serves as the cornerstone of pharmaceutical regulation in the European Union, providing a structured approach to ensuring medicinal product quality, safety, and efficacy. The framework is divided into several volumes, with Volume 4 specifically addressing Good Manufacturing Practice (GMP) for both human and veterinary medicinal products. The legal foundation for these guidelines stems from Directive 2001/83/EC, which establishes the Community code for medicinal products for human use, and Directive 2001/82/EC for veterinary medicinal products.
Within this framework, manufacturing authorization is mandatory for all pharmaceutical manufacturers in the EU, whether their products are sold within or outside the Union. Two key directives establish the principles and guidelines for GMP: Directive 2003/94/EC for human medicinal products and Directive 91/412/EEC for veterinary products. These directives are interpreted and implemented through the detailed guidelines in the Guide to Good Manufacturing Practice.
Structure and Implementation of EU Pharmaceutical Regulation
The EU pharmaceutical regulatory framework operates on multiple levels. At the highest level, EU institutions establish the legal framework through regulations and directives. EU Law includes both Regulations, which have binding legal force in every Member State, and Directives, which lay down outcomes that must be achieved while allowing each Member State some flexibility in transposing them into national laws.
The European Medicines Agency (EMA) coordinates and harmonizes at the EU level, while national regulatory authorities inspect, license, and enforce compliance locally. This multilayered approach ensures consistent quality standards while accommodating certain national considerations.
For marketing authorization, medicinal products may follow several pathways:
Authorizing body
Procedure
Scientific Assessment
Territorial scope
European Commission
Centralized
European Medicines Agency (EMA)
EU
National authorities
Mutual Recognition, Decentralized, National
National competent authorities (with possible additional assessment by EMA in case of disagreement)
EU countries concerned
This structure reflects the balance between EU-wide harmonization and national regulatory oversight in pharmaceutical manufacturing and authorization.
National Variations in Pharmaceutical Manufacturing Requirements
Austria
Austria maintains one of the more stringent interpretations of EU directives regarding Qualified Person requirements. While the EU directive 2001/83/EC establishes general qualifications for QPs, individual member states have some flexibility in implementing these requirements, and Austria has taken a particularly literal approach.
Austria also maintains a national “QP” or “eligible QP” registry, which is not a universal practice across all EU member states. This registry provides an additional layer of regulatory oversight and transparency regarding individuals qualified to certify pharmaceutical batches for release.
Denmark
Denmark has really flexible GMP certification recognition, but beyond that no real differences from Eudralex volume 4.
France
The Exploitant Status
The most distinctive feature of the French pharmaceutical regulatory framework is the “Exploitant” status, which has no equivalent in EU regulations. This status represents a significant departure from the standard European model and creates additional requirements for companies wishing to market medicinal products in France.
Under the French Public Health Code, the Exploitant is defined as “the company or organization providing the exploitation of medicinal products”. Exploitation encompasses a broad range of activities including “wholesaling or free distribution, advertising, information, pharmacovigilance, batch tracking and, where necessary, batch recall as well as any corresponding storage operations”. This status is uniquely French, as the European legal framework only recognizes three distinct positions: the Marketing Authorization Holder (MAH), the manufacturer, and the distributor.
The Exploitant status is mandatory for all companies that intend to market medicinal products in France. This requirement applies regardless of whether the product has received a standard marketing authorization or an early access authorization (previously known as Temporary Use Authorization or ATU).
To obtain and maintain Exploitant status, a company must fulfill several requirements that go beyond standard EU regulations:
The company must obtain a pharmaceutical establishment license authorized by the French National Agency for the Safety of Medicines and Health Products (ANSM).
It must employ a qualified person called a Chief Pharmaceutical Officer (Pharmacien Responsable).
It must designate a local qualified person for Pharmacovigilance.
The Pharmacien Responsable: A Unique French Pharmaceutical Role
Another distinctive feature of the French health code is the requirement for a Pharmacien Responsable (Chief Pharmaceutical Officer or CPO), a role with broader responsibilities than the “Qualified Person” defined at the European level.
According to Article L.5124-2 of the French Public Health Code, “any company operating a pharmaceutical establishment engaged in activities such as purchasing, manufacturing, marketing, importing or exporting, and wholesale distribution of pharmaceutical products must be owned by a pharmacist or managed by a company which management or general direction includes a Pharmacien Responsable”. This appointment is mandatory and serves as a prerequisite for any administrative authorization request to operate a pharmaceutical establishment in France.
The Pharmacien Responsable holds significant responsibilities and personal liability, serving as “a guarantor of the quality of the medication and the safety of the patients”. The role is deeply rooted in French pharmaceutical tradition, deriving “directly from the pharmaceutical monopoly” and applying to all pharmaceutical companies in France regardless of their activities.
The Pharmacien Responsable “primarily organizes and oversees all pharmaceutical operations (manufacturing, advertising, information dissemination, batch monitoring and recalls) and ensures that transportation conditions guarantee the proper preservation, integrity, and safety of products”. They have authority over delegated pharmacists, approve their appointments, and must be consulted regarding their departure.
The corporate mandate of the Pharmacien Responsable varies depending on the legal structure of the company, but their placement within the organizational hierarchy must clearly demonstrate their authority and responsibility. This requirement for clear placement in the company’s organization chart, with explicit mention of hierarchical links and delegations, has no direct equivalent in standard EU pharmaceutical regulations.
Germany
While Germany has many distinctive elements—including the PZN identification system, the securPharm verification approach, specialized distribution regulations, and nuanced clinical trial oversight—the GMPs from Eudralex Volume 4 are the same.
Italy
Italy has implemented a highly structured inspection system with clearly defined categories that create a distinctive national approach to GMP oversight.
National Preventive Inspections
Activating new manufacturing plants for active substances
Activating new manufacturing departments or lines
Reactivating departments that have been suspended
Authorizing manufacturing or import of new active substances (particularly sterile or biological products)
National Follow-up Inspections to verify the GMP compliance of the corrective actions declared as implemented by the manufacturing plant in the follow-up phase of a previous inspection. This structured approach to verification creates a continuous improvement cycle within the Italian regulatory system.
Extraordinary or Control Inspections: These are conducted outside normal inspection programs when necessary for public health protection.
Spain
The differences in Spain are mostly on the way an organization is registered and has no impacts on GMP operations.
Regulatory Recognition and Mutual Agreements
EU member states have received specific recognition for their GMP inspection capabilities from international partners individually.