It is crucial for a Marketing Authorization Holder (MAH) to review and approve changes made by a Contract Development and Manufacturing Organization (CDMO) for several important reasons:
Regulatory Compliance
The Market Authorization Holder (MAH) – or the sponsor for pre-commercial GMP manufacturing – bears the primary responsibility for ensuring compliance with the marketing authorization and regulatory requirements throughout the product’s lifecycle. By reviewing and approving CDMO changes, the MAH can:
Ensure changes align with the approved marketing authorization
Verify that any variations to the marketing authorization are properly submitted to regulatory authorities
Maintain oversight of post-approval change management as required by regulations
Before I go any further on the topic I want you to go and read my post Classification of Changes for GMP/GDP. This post will build on that discussion.
I think it is better for the CDMO to put a lot of thought into this, and the MAH (the client) to evaluate and adapt. For all but the big players, the volume is going to be on the CDMO’s side. But if you are the client and your CDMO hasn’t taken this into account to the appropriate degree, you need to ensure appropriate steps taken. As such the rest of this post will be written from the CDMO’s side, but the same principles apply to the MAH (and should be included in the audit program).
Remember we have three goals:
Fulfill our contractual responsibilities
Help the MAH maintain appropriate control as the product owner
Ensure alignment between both parties on change implementation
The critical requirement here is ensuring the right changes get to the right client so they can be filled the right way. Returning to basics, we are approaching changes as:
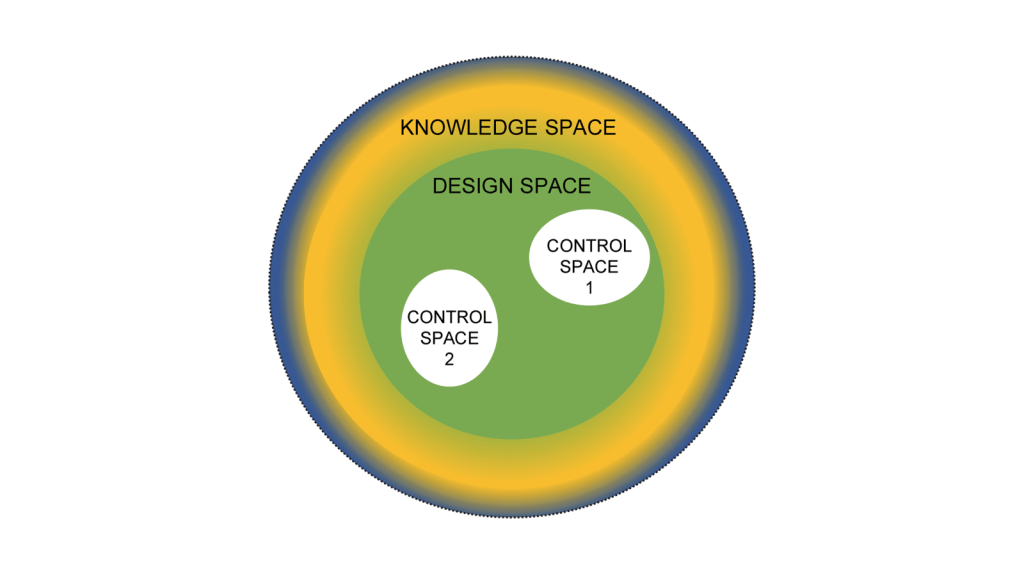
Now it’s easy to apply this to product. Create and/or receive the design space and the control space. Everything that falls into a non-established condition does not get reported to the client at time of execution. If it is “Do and Report” is is in the APQR. If it is “Do and Record” they can see it during the audit.
Where a lot of CDMOs trip up here are facility and quality system changes. My recommendation here is the same, define a design space based on the CMC section of the Common Technical Document which basically boils down to:
The CMC (Chemistry, Manufacturing, and Controls) section of a regulatory dossier typically includes the following key facility-related information:
Manufacturing Facilities
Names and addresses of all manufacturing, testing, and storage facilities involved in production
Description of the manufacturing operations performed at each site
Floor plans and layouts of production areas
Details on utilities and support systems (HVAC, water, gases, etc.)
Information on facility design features for contamination control and product protection
Equipment
List of major production and laboratory equipment
Equipment specifications and capacities
Cleaning and maintenance procedures for equipment
Environmental Controls
Description of clean room classifications and environmental monitoring programs
Air handling systems and controls
Water systems (purified water, water for injection) and controls
Material Flow
Personnel and material flow diagrams
Segregation of operations to prevent cross-contamination
Quality Control Laboratories
Description of QC lab facilities and equipment
Environmental controls in QC labs
Storage Areas
Description of storage facilities for raw materials, intermediates, and finished products
Storage conditions and controls (temperature, humidity, etc.)
There is a whole lot of wiggle room here in things that fall into “Do and Record.” By building this into your change control system you can delineate what goes to to the client and what doesn’t. I recommend sitting down with this list and deciding what types of changes fall into “Tell and Do” – what you ask permission from clients before doing; “Do and Report” – what goes in the APQR; and, “Do and Record” – what the client sees when they audit.
You know have good rules on what changes go to a client for prior approval and which ones do not. This gets codified in two places: the change control process and the quality/technical agreement.
Some other things to build into your change control process:
Documenting when a client requests a change, the reason and the impact on the platform. Remember you have other clients, and more and more CDMO’s are offering a platform, so there needs to be appropriate review and endorsement.
Think through how changes to facility (and other platform elements) are communicated and gated for multiple clients. Have a mechanism to manage client specific activities and to track first-product impacted for multiple products.
Have clear timelines and expectations on change communication and approval with the client in the quality/technical agreement. Hold each other accountable.
Have contingency plans. There will always be that one client who will be in shortage if you make that urgent change just when you want/need to.
Have a method for evaluating requested changes to the change plan by clients and making decisions around it. There will be that one client who doesn’t agree or wants something weird that disagrees with what all the other clients want.
Have rules in place to manage changes inactive for long periods or extensions specific for those changes that rise to client approval. These will have a different flow than internal changes.
I’ve used a bit of commercial headspace for this post, relying on the APQR. For clinical processes, product tends to fall into campaign-mindset, so “Do and Report” ends up being more a clinical campaign change report than an APQR.
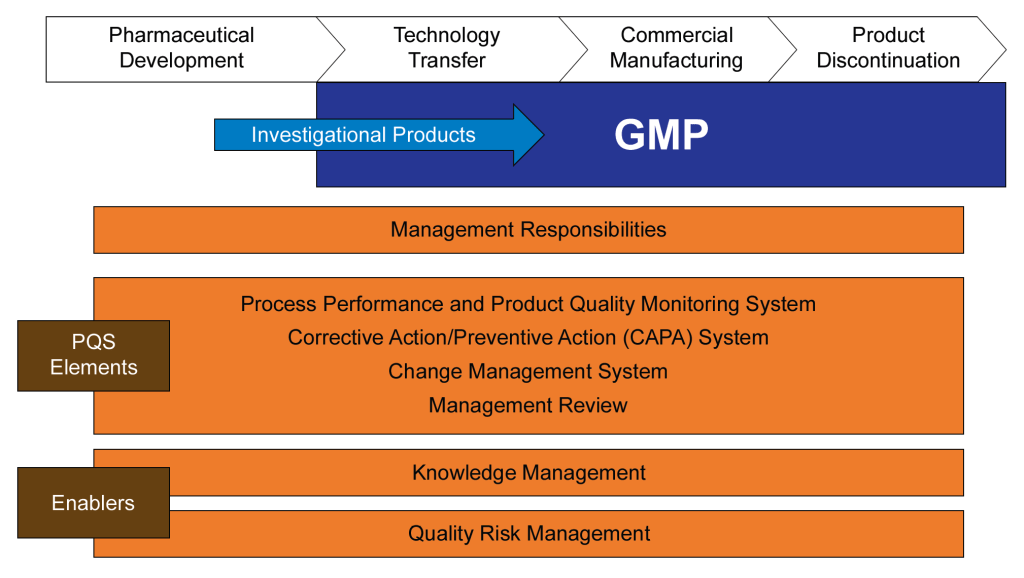
The International Conference on Harmonization (ICH) was established to harmonize the technical requirements for pharmaceutical product registration across Europe, Japan, and the United States. ICH Q10, finalized in June 2008, emerged from this initiative as a guideline for a comprehensive Pharmaceutical Quality System (PQS) applicable throughout the product lifecycle. It was adopted by the FDA in April 2009, following its implementation by the European Commission in July 2008.
ICH Q10 aims to provide a model for pharmaceutical manufacturers to develop and maintain effective quality management systems. The guideline emphasizes a lifecycle approach, integrating quality management principles from ISO standards and regional GMP requirements. The primary objectives of ICH Q10 include:
Ensuring consistent product quality that meets customer and regulatory requirements.
Establishing effective monitoring and control systems for process performance and product quality.
Promoting continual improvement and innovation throughout the product lifecycle.
The guideline outlines the key elements of management responsibilities, Corrective and Preventive Action (CAPA) , process performance and product quality monitoring, change management, and management review. ICH Q10 is usually considered part of the “Quality Trio” with ICH Q8 and Q9. Quality by design is only possible through proper risk management and a robust quality system.
FDA Guidance for Industry on Quality Systems Approach to Pharmaceutical CGMP Regulation
The FDA developed guidance on implementing modern quality systems and risk management practices to align with the CGMP (Current Good Manufacturing Practice) requirements outlined in parts 210 and 211 of the FDA regulations. These regulations govern the manufacturing of human and veterinary drugs, including biological products. Published in 2006, this guidance should be viewed as part of a continuum of thought with ICH Q10 and not as an earlier draft.
This guidance aims to assist manufacturers in meeting cGMP requirements by adopting a comprehensive quality systems model. It emphasizes the integration of quality systems with regulatory requirements to ensure full compliance without imposing new expectations on manufacturers. Key aspects of the guidance include:
Highlighting the consistency of the quality systems model with cGMP regulations.
Encouraging the use of risk management and quality systems to enhance compliance and product quality.
Providing a framework for manufacturers to gain control over their manufacturing processes.
Six-System Inspection Model
The FDA’s Six-System Inspection Model is a framework introduced in this guidance to ensure compliance with current Good Manufacturing Practice (CGMP) regulations in the pharmaceutical industry. This model helps FDA inspectors evaluate the robustness of a company’s quality management system by focusing on six key subsystems.
I am a huge fan of the six subsystem approach. Basically we have here the organization of the quality manual, a guide to what standards you need to write in a bigger company, and a franework for understanding the cGMPs as a whole (great for education purposes).
Here’s a detailed explanation of each subsystem:
1. Quality System
Role: Acts as the central hub for all other systems, ensuring overall quality management.
Focus: Management responsibilities, internal audits, CAPA (Corrective and Preventive Actions), and continuous improvement.
Importance: Ensures that all other systems are effectively integrated and managed to maintain product quality and regulatory compliance.
2. Facilities and Equipment System
Role: Ensures that facilities and equipment are suitable for their intended use and maintained properly.
Focus: Design, maintenance, cleaning, and calibration of facilities and equipment.
Importance: Prevents contamination and ensures consistent manufacturing conditions.
3. Materials System
Role: Manages the control of raw materials, components, and packaging materials.
Focus: Supplier qualification, receipt, storage, inventory control, and testing of materials.
Importance: Ensures that only high-quality materials are used in the manufacturing process, reducing the risk of product defects.
4. Production System
Role: Oversees the actual manufacturing processes.
Focus: Process controls, batch records, in-process controls, and validation.
Importance: Ensures that products are manufactured consistently and meet predefined quality criteria.
5. Packaging and Labeling System
Role: Manages the packaging and labeling processes to ensure correct and compliant product presentation.
Focus: Label control, packaging operations, and labeling verification.
Importance: Prevents mix-ups and ensures that products are correctly identified and used.
6. Laboratory Controls System
Role: Ensures the reliability of laboratory testing and data integrity.
Focus: Sampling, testing, analytical method validation, and laboratory records.
Importance: Verifies that products meet quality specifications before release.
Integration and Interdependence
Quality System as the Fulcrum: The quality system is the central element that integrates all other subsystems. It ensures that each subsystem functions correctly and is aligned with overall quality objectives.
State of Control: The primary goal of the six-system inspection model is to ensure that each subsystem is in a state of control, meaning it operates within predefined limits and consistently produces the desired outcomes.
The Six-System Inspection Model provides a structured approach for FDA inspectors to assess the compliance and effectiveness of a pharmaceutical company’s quality management system. By focusing on these six subsystems, the FDA ensures that all aspects of manufacturing, from raw materials to final product testing, are adequately controlled and managed to maintain high standards of product quality and safety.
A Complementary and Holistic Approach
Both ICH Q10 and the FDA’s guidance on quality systems approach aim to enhance the quality and safety of pharmaceutical products through robust quality management systems. ICH Q10 provides a harmonized model applicable across the product lifecycle, while the FDA guidance focuses on integrating quality systems with existing CGMP regulations. Together, they support the pharmaceutical industry in achieving consistent product quality and regulatory compliance.
Aspect
ICH Q10
FDA Guidance on CGMP
ISO 13485 and 21 CFR 820
ISO 9000
Purpose and Scope
Comprehensive model for pharmaceutical quality systems across the product lifecycle.
Quality systems approach to ensure CGMP compliance in pharmaceuticals.
Quality management system for medical devices, incorporating ISO 13485 and regulatory requirements of 21 CFR 820.
Fundamentals and vocabulary for quality management systems applicable to any industry.
Industry Focus
Specifically for the pharmaceutical industry.
Specifically for the pharmaceutical industry.
Specifically for the medical device industry.
Applicable to any industry.
Key Elements
Management responsibilities, CAPA, process performance, change management, management review.
Management responsibilities, quality systems, process validation, continuous improvement.
Quality management principles, terms, and definitions.
Regulatory Focus
Strong emphasis on regulatory compliance and lifecycle management.
Strong emphasis on regulatory compliance with CGMP.
Incorporates regulatory requirements specific to medical devices (21 CFR 820).
Does not directly address regulatory compliance.
Flexibility
Flexible, adaptable to specific product and process needs.
More prescriptive with specific compliance requirements.
Harmonized with international standards but includes specific regulatory requirements.
Provides a broad framework for customization.
Management Involvement
Emphasizes management’s role in quality and regulatory compliance.
Emphasizes management’s role in quality and CGMP integration.
Emphasizes management’s role in quality and risk-based decision making.
Emphasizes management’s role in quality and customer satisfaction.
Implementation
Tailored to pharmaceutical manufacturing, integrating quality management principles.
Mandates oversight and controls over drug manufacturing processes.
Requires a quality manual and specific documentation practices; aligned with international standards.
Requires customization to specific industry needs.
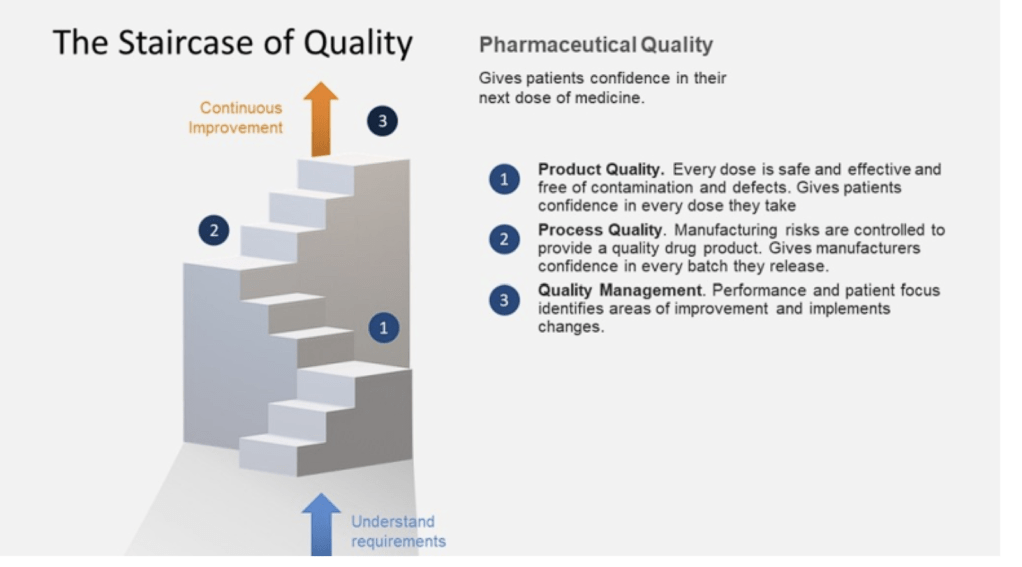
These two documents were developed at the same time and represents the thinking twenty years ago in laying down an approach that still matters today. I usually regard the six system approach as a deepening and defining of what Q10 means by process performance and product quality monitoring.
What is the current agency thinking?
The FDA and other revulatory agencies haven’t stopped their thinking in 2008. Sixteen years later we see the continued push for quality culture and quality maturity. The FDA continues to make this a top priority, as we’ve been seeing in their annual drug shortage reports to Congress. There are a few themes we continue to see driven home.
The Patient is the Customer
Quality management must be customer-focused, ensuring that all processes and materials meet their intended use. Senior management’s commitment is crucial for a strong QMS, which emphasizes proactive quality assurance over reactive quality control. Robust supplier relationships and oversight programs are essential to manage variability in materials and processes.
This application of a core priciple in ISO 9000 may seem to basic to some, but I think it is central to a lot of messaging and should never be taken for granted.
Benefits of Better Quality Performance
A continued focus that a quality-focused culture leads to:
Early problem detection
Enhanced process stability and productivity
Fewer major deviations and failures
Efficient QA release of batches
Reduced customer complaints and returns
Protection of brand and competitiveness
Management Oversight of Drug Quality
Management must address sources of variability, including people, materials, methods, measurements, machines, and environment. Risk management should be dynamic and ongoing, facilitating continual learning and improvement.
Corrective Action and Preventive Action (CAPA)
A structured approach to investigating complaints, product rejections, nonconformances, recalls, deviations, audits, regulatory inspections, and trends is essential. CAPA should determine root causes and implement corrective actions.
Change Management
Timely and effective change management ensures corrections and improvements are undertaken efficiently. This includes implementing product quality improvements, process improvements, variability reduction, innovations, and pharmaceutical quality system enhancements.
Management Review
Management is responsible for quality policy, QMS effectiveness, internal communications, resource management, and supply chain oversight. This includes ensuring the quality of incoming materials and outsourced activities.
Quality Culture Driven by Top Management
A strong corporate quality culture is driven by daily decisions and executive oversight. Sustainable compliance requires aiming for high standards rather than just meeting minimum requirements. Quality management maturity involves proactive and preventive actions, iterative learning, and leveraging modern technologies.
Facility Lifecycle
Senior management must ensure the suitability of operational design, control, and maintenance. This includes addressing infrastructure reliability, appropriateness for new product demands, and mitigating equipment/facility degradation.
Risk Management in Manufacturing
Human factors and manual interventions pose significant risks in pharmaceutical manufacturing. Automation and separation technologies can mitigate these risks, but many facilities still rely on manually intensive processes. Leveraging new technologies and practices is a huge opportunity.
This approach is reflected in the FDA’s Quality Management Maturity (QMM), which promotes advanced quality management practices within drug manufacturing establishments.
Goals of the QMM Program
Foster a Strong Quality Culture Mindset: Encourage establishments to integrate quality deeply into their organizational culture.
Recognize Advanced Quality Management Practices: Acknowledge and reward establishments that go beyond basic CGMP (Current Good Manufacturing Practices) requirements.
Identify Growth Opportunities: Provide suggestions for enhancing quality management practices.
Minimize Risks to Product Availability: Ensure a reliable market supply by reducing quality-related failures and maintaining performance during supply chain disruptions.
Key Components of the QMM Program
Management Commitment to Quality: Leadership must prioritize quality, set clear objectives, and integrate these with business goals. Effective management review processes are crucial.
Business Continuity: Establishments should develop robust plans to handle disruptions, ensuring consistent operations and supply chain reliability.
Advanced Pharmaceutical Quality System (PQS): Implementing quality principles like Quality by Design (QbD) and risk management approaches to maintain system reliability and minimize production disruptions.
Technical Excellence: Emphasizing data management, innovative manufacturing processes, and advanced technologies to enhance quality and operational efficiency.
Employee Engagement and Empowerment: Encouraging employees to take ownership of quality, make suggestions, and understand their impact on product quality and patient safety.
Implementation and Assessment
The FDA has developed a prototype assessment protocol to evaluate QMM. This includes a standardized approach to minimize bias and ensure objectivity. Someday, eventually, it will move away from constant prototyping.
Assessments will focus on qualitative aspects, such as the establishment’s quality culture and how it uses data to drive improvements.
Benefits of QMM
Enhanced Supply Chain Reliability: By adopting mature quality management practices, establishments can reduce the occurrence of quality-related failures. The fact shortages continue to be so damning to our industry is a huge wake-up call.
Proactive Continual Improvement: Encourages a proactive approach to quality management, leveraging technological advancements and integrated business operations.
Long-term Cost Savings: Investing in a mature quality culture can lead to fewer compliance issues, reduced inspection needs, and overall cost reductions.
Conclusion
The FDA’s QMM program aims to transform how pharmaceutical quality is perceived, measured, and rewarded. The program seeks to ensure a more reliable drug supply and better patient outcomes by fostering a strong quality culture and recognizing advanced practices. It should be seen as part of a 20-year commitment from the agency in alignment with its international partners.
Classification of change controls within change management is a common and widely accepted best practice. It stems from the requirement that change proposals as assessed from a risk perspective, where:
the level of rigor, effort and documentation is commensurate with the level of risk,
the risk assessments adequately evaluate the potential risks and benefits of changes to product quality, safety and efficacy, and
those risk assessments consider the potential risks and benefits to other products, processes and systems.
Classification for GMP/GDP changes itself is not a requirement, it is a guidance, best found in the PIC/S Recommendation “How to Evaluate and Demonstrate the Effectiveness of a Pharmaceutical Quality System in relation to Risk-based Change Management” (PI 054-1) which states in section 5.2 “Change Management procedures often require a risk-based classification (e.g. critical, major, minor) to be assigned to proposed changes as well as an impact assessment to be performed. The latter routinely determines the potential impacts of the proposed change on various items, such as product quality, documentation, cleaning, maintenance, regulatory compliance, etc. In some cases, especially for simple and minor/low risk changes, an impact assessment is sufficient to document the risk-based rationale for a change without the use of more formal risk assessment tools or approaches.”
The PIC/S tells us that these categories drive the amount of rigor a change control requires, which is a great reason to have them. We spend time creating and confirming our categories, and then we only need to perform more rigorous risk assessments on the big changes.
How should we build this risk-based classification system? There are four criteria that drive this:
Potential regulatory impact
Potential impact on the qualified and validated state
Potential impact on the ability to disposition and ship product
Complexity
I tend to use only two categories, defined like this:
Major has Significant Impact: Changes that have a considerable potential impact on the process, product quality, safety, or regulatory status.
Minor has Limited Impact: Changes that have minimal or no significant impact on the process, product quality, safety, or regulatory status.
For regulatory impact, it really is as easy as dividing things into the four categories. “Do, Report, and Do and Record are minors. “Do and Tell” are majors, and “Tell and Do are either majors or critical based on how you slice it.
When considering potential validation impact you’ll leverage your process risk assessments and your validated state to determine what is in that bucket. This is why I like a document like an operational control strategy because this tells me exactly what impacts my validated state and I can just it to form this category.
The potential impact on the ability to disposition and ship the product has me looking at what can impact the ability to release and get the product out the door, which is an important aspect of what we do. Remember, a shortage of products is a quality issue.
Complexity looks at how many processes and systems are impacted and how many functions and areas are involved. The more complex, the more formal risk assessment is required. For example, you might use groupings like this:
Low level of complexity
Requires actions from the change owner and the system owner’s department(s) only
Impacts 1 system
<10 document revisions (approximate)
<2 potential training audiences (approximate)
Higher complexity:
Requires actions from more than change owner and system owner
Impacts more than one system
>10 document revisions
>2 potential training audiences (approximate)
The where of making the classification also makes a difference. I recommend up front, agreed to by the change owner and quality and it then drives everything. Doing it just before approval really just decides who gets to approve the change control and whether it goes to CCRB or not.
These classifications can be loose guidelines; for example, a table that looks at the first three categories and then by complexity. Your rating depends on whichever Impact or Complexity is higher.
Impact of Change (regulatory, validation, product)
Complexity of Change
Minor
No risk to patient as assessed by SISPQ, product, or validated equipment or process AND No regulatory impact.
Limited impact to only one system/functional area AND Has defined process for implementation of change. (e.g. all action items are per defined procedures)
Major
Potential impact to patient or product SISPQ or validated equipment or process or compliance
Impacts multiple systems / functional areas OR Has defined process for implementation of change
Critical
High likelihood of impact to patient, product SISPQ or validated equipment or process or compliance
Impacts multiple systems / functional areas OR Implementation activities are not pre-defined or governed by formal internal system
Or we could try for something much more specific. The advantage of specific is any change owner can start making the determination. Something like this:
Change Category
Change Description
Manufacturing Processes
In-process labeling
Changes to Process Control and Operating Parameters (tightening/shifting) within current batch record (does not impact established conditions)
The addition of in-process or final product samples
Changes to sample volume for in-process or finished product samples
Addition of new ancillary equipment (e.g. no product contact, does not control process steps) to the process
Analytical Methods
Changes to the qualification of a critical reagent (i.e., in-house produced assay standards and controls)
Use of an additional new instrument of the identical model and vendor
Change in compendial method to comply with formal updates to compendia, provided it does not involve the widening of system suitability or acceptance criteria
Equipment/instruments calibration, maintenance, and cleaning
Changes to software or validated analytical spreadsheets that do not impact the current validated state of the method
Movement of instruments from one location to another in the same room/lab
Initial validation of analytical spreadsheets for use in calculation of data and results defined by a specific analytical method, provided it does not replace a worksheet in an SOP (if so, this change may be reportable)
Changes to non-critical equipment or materials that allow “or equivalent” in current method, provided method re-validation is not required
Drug Substance or Drug Product Specifications/ Limits
Changes to the sampling plan involving changes to the number of extra samples or amount of sample provided to QC or CMO as appropriate.
Changes to the storage and/or shipping conditions of samples (except for stability vials)
Raw Materials/Com ponents
Compendial Specification Changes to meet Compendial updates
Non-product contact filters
Vendor increase or decrease in the number of items per shipping container, or the size of the shipping or outer container
Changes to the vendor Certificate of Analysis (format change only)
Changes in recommended expiration date and/or storage conditions of raw material
Finished Goods
Catalog Number changes to components
Creation of label at contract manufacturing site for existing presentation (assuming ‘No’ other change to already approved label)
Changing position of pharmacode on leaflet
Computer
When there is no validation impact
Facility, Utilities, Systems and Equipment (including Automation)
Equipment/instrument maintenance
Decommissioning of equipment not classified as critical equipment
Computer programming that affects non-production equipment
Alarms (i.e., notification system for out of tolerances)
Cleaning and Sanitization of Manufacturing facilities and non-product Contact equipment
Upgrade of Application Software or operating system
Alarm set point changes
Creating user groups and modifying user group privileges
Tuning parameter, adjustment to the gain, reset and rate of a PID controller
Phase or sequence change that does not affect the function and performance
Modifying a phase prompt or message (technical change)
Addition of a graphic, adding or changing a non-static device to a graphic (technical change)
Addition or changing to an interlock/permissive trigger
Changes to alarm paging/notification functionality
Spend the time on your classification structure. You will use it to:
Determine level of risk assessment (major yes, minor no)
Determine approvals (minors can be as simple as change owner and quality)
Does this change require a CCRB? Only send majors.
As leaders, embracing change, both the ones we foster and change that stems from other places within and without our organizations, is critical. By embracing change ourselves, we lead by example and demonstrate the behaviors and mindset they expect from their teams. This can create a ripple effect, encouraging others to adopt a similar attitude toward change.
Understanding the Importance of Change
Recognize the Necessity of Change: Change is inevitable and essential for growth and improvement. Leaders who embrace change are more adaptable and capable of handling various challenges.
View Change as an Opportunity: Change opens doors to new opportunities, skills, and knowledge. It fosters innovation and can lead to excellence by pushing leaders and their teams out of their comfort zones.
Developing Key Leadership Skills
Adaptability: Being adaptable allows leaders to act quickly, face conflicts head-on, and learn from failures. This skill is pivotal in managing and leading change successfully.
Visionary Thinking: Setting a clear direction and purpose for the future helps inspire others to embrace change. Visionary leaders can motivate their teams by outlining long-term strategies and goals.
Communication and Influencing: Effective communication is crucial during times of change. Leaders should clearly articulate what changes are occurring, why they are necessary, and how they will be implemented. Listening with empathy and being transparent helps build trust and engagement.
Emotional Intelligence: It is essential to manage one’s emotions and respond well to others’ emotions. Recognizing and acknowledging others’ feelings can help mitigate stress and resistance to change.
Resilience and Persistence: Change can be challenging and unpredictable. Resilient leaders can bounce back from obstacles and remain focused on desired outcomes. Persistence helps sustain momentum throughout the change process.
Practical Steps to Embrace Change
Build a Support System: Don’t go it alone. Seek support from mentors, peers, and team members. Encourage your employees to do the same.
Create a Clear Vision and Plan: Establish and communicate a vision for the change early on. Develop a comprehensive change management plan that includes clear communication channels and methods to monitor progress.
Model Expected Behaviors: Demonstrate the behaviors you expect from your team. Show a willingness to try new things, ask questions, and share insights about the change process.
Engage and Support Employees: Regularly share information about the status and impact of the change. Show empathy and provide opportunities for employees to voice their concerns and successes.
Recognize and Celebrate Successes: Acknowledge and celebrate small victories along the way. This helps maintain motivation and reinforces positive behaviors.
Be Patient and Understanding: Understand that some employees may adapt more quickly than others. Provide ongoing support and check-ins to ensure everyone is coping well with the change.
Leading by Example
Embrace a Proactive Attitude: Be proactive rather than reactive. Seek out new opportunities and challenges, and constantly look for ways to improve and innovate.
Show Humility and Openness: Foster trust and psychological safety by being humble, authentic, and open. This enables your team to reach their full potential and navigate changes effectively.
Encourage Leadership at All Levels: Empower your team members to take on leadership roles and make decisions. This helps build a change-ready culture where everyone is involved in the process.
Encouraging your team to embrace change involves clear communication, active involvement, and supportive leadership.
Understand and Address Resistance
Identify the Root Causes of Resistance: Understand why team members might resist change. Common reasons include fear of the unknown, lack of trust, loss of control, and attachment to the status quo. You can address these issues more effectively by listening to their concerns and empathizing with their emotions.
Communicate the Vision and Benefits: Explain why the change is necessary, the expected outcomes, and how it will benefit the team and the organization. Use stories, examples, and testimonials to illustrate the benefits and inspire the team.
Involve and Empower Your Team
Encourage Participation: Involve team members in the decision-making process. Seek their input, feedback, and suggestions on implementing the change. This will help them feel valued and give them a sense of ownership over the change process.
Provide Training and Support: Offer training and resources to help team members adjust to the change and ensure they have the skills and knowledge to succeed in the new environment.
Create a Supportive Environment: Foster a culture of open communication where team members feel comfortable sharing their ideas and concerns. This can help build trust and reduce resistance.
Communicate Effectively
Be Clear and Transparent: Communicate clearly and consistently about the change. Explain the change’s purpose, scope, and impact and how it aligns with the organization’s vision and goals.
Tailor Your Communication: Different stakeholders may react and be concerned about the change. Tailor your communication to address their specific needs and interests.
Use Multiple Channels: Use various communication methods to reach all team members. This can include team meetings, one-on-one sessions, emails, and interactive platforms.
Foster a Change-Ready Culture
Promote a Culture of Continuous Improvement: Encourage a mindset of adaptability and continuous learning. This helps team members see change as a natural part of growth and improvement.
Build Trust and Collaboration: Foster a culture of trust and collaboration where team members feel supported and valued. This can help reduce resistance and increase engagement with the change process.
ASTM E2500, the Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, is intended to “satisfy international regulatory expectations in ensuring that manufacturing systems and equipment are fit for the intended use and to satisfy requirements for design, installation, operation, and performance.”
The ASTM E2500 approach is a comprehensive framework for specification setting, design, and verification of pharmaceutical and biopharmaceutical manufacturing systems and equipment. It emphasizes a risk- and science-based methodology to ensure that systems are fit for their intended use, ultimately aiming to enhance product quality and patient safety.
Despite its 17-year history, it is fair to say it is not the best-implemented standard. There are still many unrealized opportunities and some major challenges. I don’t think a single organization I’ve been in has fully aligned, and ASTM E2500 can feel aspirational.
Key Principles
Risk Management: The approach integrates risk management principles from ICH Q8, Q9, and Q10, focusing on identifying and mitigating risks to product quality and patient safety throughout the lifecycle of the manufacturing system.
Good Engineering Practices (GEP): It incorporates GEP to ensure systems are correctly designed, installed, and operated.
Flexibility and Efficiency: It strives for a more flexible and efficient organization of verification activities that can be adapted to each project’s specific context.
Regulatory agencies expect drugmakers to persuade them that we know our processes and that our facilities, equipment, systems, utilities, and procedures have been established based on concrete data and a thorough risk assessment. The ASTM E2500 standard provides a means of demonstrating that all of these factors have been validated in consideration of carefully evaluated risks.
What the Standard Calls for
Four Main Steps
Requirements: Define the system’s needs and critical aspects. Subject Matter Experts (SMEs) play a crucial role in this phase by defining needs, identifying critical aspects, and developing the verification strategy.
Specification & Design: Develop detailed specifications and design the system to meet the requirements. This step involves thorough design reviews and risk assessments to ensure the system functions as intended.
Verification: Conduct verification activities to confirm that the system meets all specified requirements. This step replaces the traditional FAT/SAT/IQ/OQ/PQ sequence with a more streamlined verification process that can be tailored to the project’s needs.
Acceptance & Release: Finalize the verification process and release the system for operational use. This step includes the final review and approval of all verification activities and documentation.
Four Cross-Functional Processes
Good Engineering Practices (GEP): Ensure all engineering activities adhere to industry standards and best practices.
Quality Risk Management: Continuously assess and manage risks to product quality and patient safety throughout the project.
Design Review: Regularly reviews the system design to ensure it meets all requirements and addresses identified risks.
Change Management: Implement a structured process for managing system changes to ensure that all modifications are appropriately evaluated and documented.
Applications and Benefits
Applicability: The ASTM E2500 approach can be applied to new and existing manufacturing systems, including laboratory, information, and medical device manufacturing systems.
Lifecycle Coverage: It applies throughout the manufacturing system’s lifecycle, from concept to retirement.
Regulatory Compliance: The approach is designed to conform with FDA, EU, and other international regulations, ensuring that systems are qualified and meet all regulatory expectations.
Efficiency and Cost Management: By focusing on critical aspects and leveraging risk management tools, the ASTM E2500 approach can streamline project execution, reduce time to market, and optimize resource utilization.
The ASTM E2500 approach provides a structured, risk-based framework for specifying, designing, and verifying pharmaceutical and biopharmaceutical manufacturing systems. It emphasizes flexibility, efficiency, and regulatory compliance, making it a valuable tool for ensuring product quality and patient safety.
What Makes it Different?
ASTM E2500
The more traditional approach
Testing Approach
It emphasizes a risk-based approach, focusing on identifying and managing risks to product quality and patient safety throughout the manufacturing system’s lifecycle. This approach allows for flexibility in organizing verification activities based on the specific context and critical aspects of the system.
Typically follows a prescriptive sequence of tests (FAT, SAT, IQ, OQ, PQ) as outlined in guidelines like EU GMP Annex 15. This method is more rigid and less adaptable to the specific needs and risks of each project.
Verification vs Qualification
The term “verification” encompasses all testing activities, which can be organized more freely and rationally to optimize efficiency. Verification activities are tailored to the project’s needs and focus on critical aspects.
Follows a structured qualification process (Installation Qualification, Operational Qualification, Performance Qualification) with predefined steps and documentation requirements.
Role of Subject Matter Experts
SMEs play a crucial role from the start of the project, contributing to the definition of needs, identification of critical aspects, system design review, and development of the verification strategy. They are involved throughout the project lifecycle.
SMEs are typically involved at specific points in the project lifecycle, primarily during the qualification phases, and may not have as continuous a role as in the ASTM E2500 approach.
Integration of Good Engineering Practices
Offers greater flexibility in organizing verification activities, allowing for a more efficient and streamlined process. This can lead to reduced time to market and optimized resource utilization.
While GEP is also important, the focus is more on the qualification steps rather than integrating GEP throughout the entire project lifecycle.
Change Management
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
Documentation
Documentation is focused on risk management and verification activities, ensuring compliance with international regulations (FDA, EU, ICH Q8, Q9, Q10). The approach is designed to meet regulatory expectations while allowing for flexibility in documentation.
quires extensive documentation for each qualification step, which can be more cumbersome and less adaptable to specific project needs.
Opinion
I’m watching to see what the upcoming update to Annex 15 will do to address the difficulties some see between an ATSM E2500 approach and the European regulations. I also hope we will see an update to ISPE Baseline® Guide Volume 5: Commissioning and Qualification to align an approach.
ISPE Baseline® Guide Volume 5
ATSM E2500
Design inputs Impact assessment Design Qualification Commissioning Multiple trial runs to get things right IQ, OQ, PQ, and acceptance criteria GEP Scope and QA Scope overlapped Focused on Documentation Deliverables Change Management
Design inputs Design Review Risk Mitigation Critical Control Parameters define Acceptance Criteria Verification Testing Performance Testing GEP Scope and QA Scope have a clear boundary Process, Product Quality and Patient Safety Quality by Design, Design Space, and Continuous Improvement
To be honest I don’t think ATSM E2500, ISPE Guide 5, or anything else has the balance just right. And your program ends up being a triangulation between these and the regulations. And don’t even bring in trying to align GAMP5 or USP <1058> or…or…or…
And yes, I do consider this part of my 3-year plan. I look forward to the challenges of a culture shift, increased SME involvement, formalization of GEPs (and teaching engineers how to write), effective change management, timely risk assessments, and comprehensive implementation planning.