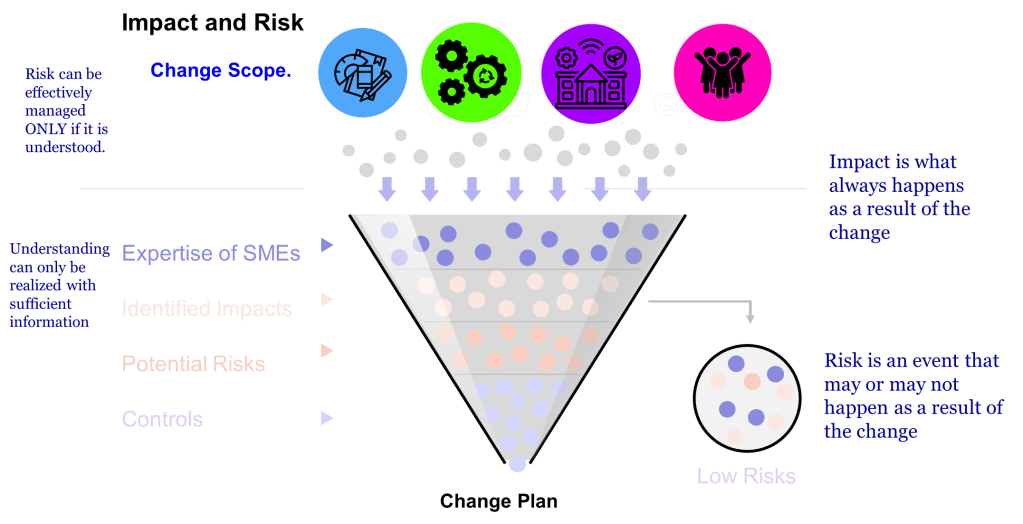
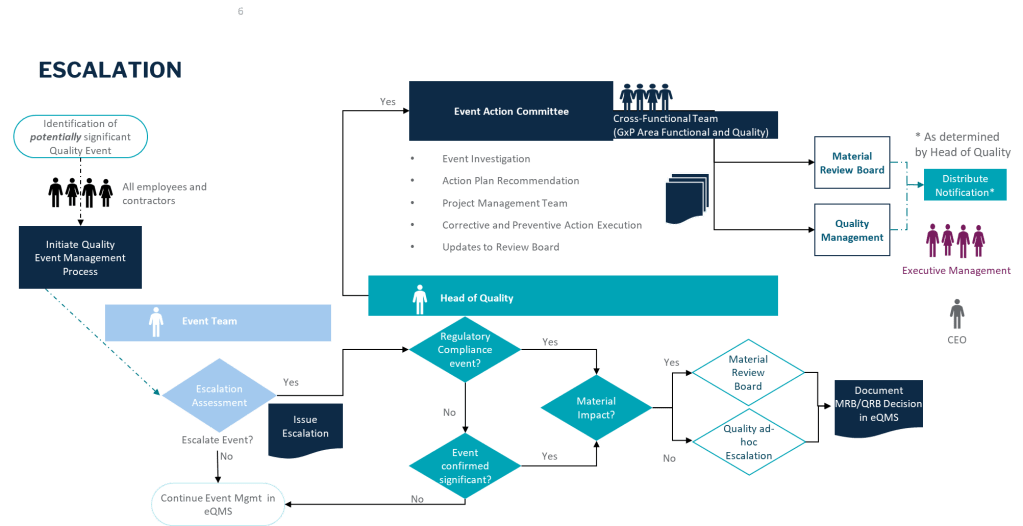
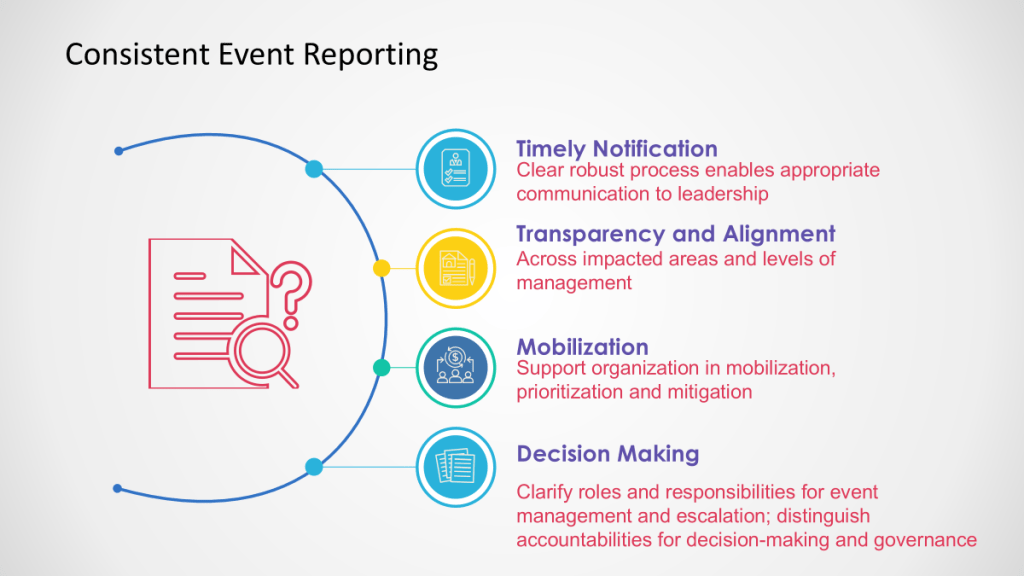
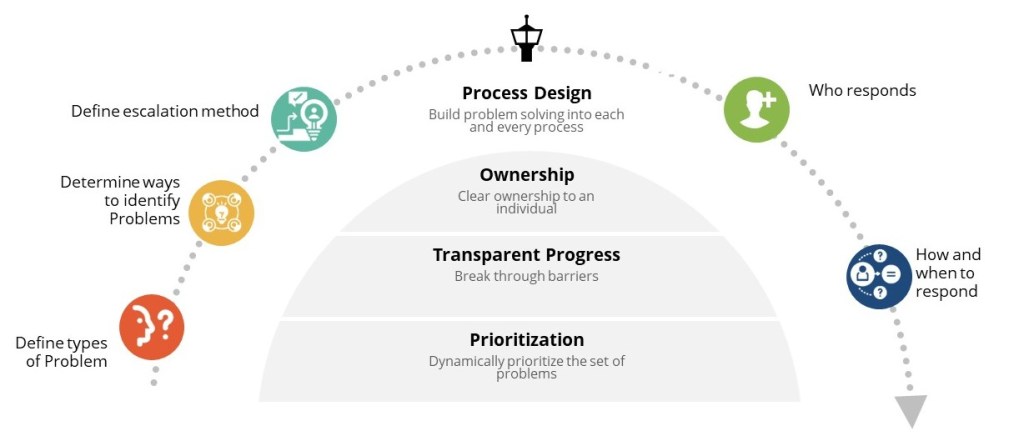
If you are like me, you face a fundamental choice on a daily (or hourly basis): we can either develop distributed decision-making capability throughout our organizations, or we can create bottlenecks that compromise our ability to respond effectively to quality events, regulatory changes, and operational challenges. The reactive control mindset—where senior quality leaders feel compelled to personally approve every decision—creates dangerous delays in an industry where timing can directly impact patient safety.
It makes sense, we are an experience based profession, so decisions tend to need by more experienced people. But that can really lead to an over tendency to make decisions. Next time you are being asked to make a decision as these four questions.
1. Who is Closest to the Action?
Proximity is a form of expertise. The quality team member completing batch record reviews has direct insight into manufacturing anomalies that executive summaries cannot capture. The QC analyst performing environmental monitoring understands contamination patterns that dashboards obscure. The validation specialist working on equipment qualification sees risk factors that organizational charts miss.
Consider routine decisions about cleanroom environmental monitoring deviations. The microbiologist analyzing the data understands the contamination context, seasonal patterns, and process-specific risk factors better than any senior leader reviewing summary reports. When properly trained and given clear escalation criteria, they can make faster, more scientifically grounded decisions about investigation scope and corrective actions.
2. Pattern Recognition and Systematization
Quality systems are rich with pattern decisions—deviation classifications, supplier audit findings, cleaning validation deviations, or analytical method deviations. These decisions often follow established precedent and can be systematized through clear criteria derived from your quality risk management framework.
This connects directly to ICH Q9(R1)’s principle of formality in quality risk management. The level of delegation should be commensurate with the risk level, but routine decisions with established precedent and clear acceptance criteria represent prime candidates for systematic delegation.
3. Leveraging Specialized Expertise
In pharmaceutical quality, technical depth often trumps hierarchical position in decision quality. The microbiologist analyzing contamination events may have specialized knowledge that outweighs organizational seniority. The specialist tracking FDA guidance may see compliance implications that escape broader quality leadership attention.
Consider biologics manufacturing decisions where process characterization data must inform manufacturing parameters. The bioprocess engineer analyzing cell culture performance data possesses specialized insight that generic quality management cannot match. When decision authority is properly structured, these technical experts can make more informed decisions about process adjustments within validated ranges.
4. Eliminating Decision Bottlenecks
Quality systems are particularly vulnerable to momentum-stalling bottlenecks. CAPA timelines extend, investigations languish, and validation activities await approvals because decision authority remains unclear. In our regulated environment, the risk isn’t just a suboptimal decision—it’s often no decision at all, which can create far greater compliance and patient safety risks.
Contamination control strategies, environmental monitoring programs, and cleaning validation protocols all suffer when every decision must flow through senior quality leadership. Strategic delegation creates clear authority for qualified team members to act within defined parameters while maintaining appropriate oversight.
Building Decision Architecture in Quality Systems
Effective delegation in pharmaceutical quality requires systematic implementation:
Phase 1: Decision Mapping and Risk Assessment
Using quality risk management principles, catalog your current decision types:
- High-risk, infrequent decisions: Major CAPA approvals, manufacturing process changes, regulatory submission decisions (retain centralized authority)
- Medium-risk, pattern decisions: Routine deviation investigations, supplier performance assessments, analytical method variations (candidates for structured delegation)
- Low-risk, high-frequency decisions: Environmental monitoring trend reviews, routine calibration approvals, standard training completions (ideal for delegation)
Phase 2: Competency-Based Authority Matrix
Develop decision authority levels tied to demonstrated competencies rather than just organizational hierarchy. This should include:
- Technical qualifications required for specific decision categories
- Experience thresholds for handling various risk levels
- Training requirements for expanded decision authority
- Documentation standards for delegated decisions
Phase 3: Oversight Evolution
Transition from pre-decision approval to post-decision coaching. This requires:
- Quality metrics tracking decision effectiveness across the organization
- Regular review of delegated decisions for continuous improvement
- Feedback systems that support decision-making development
- Clear escalation pathways for complex situations