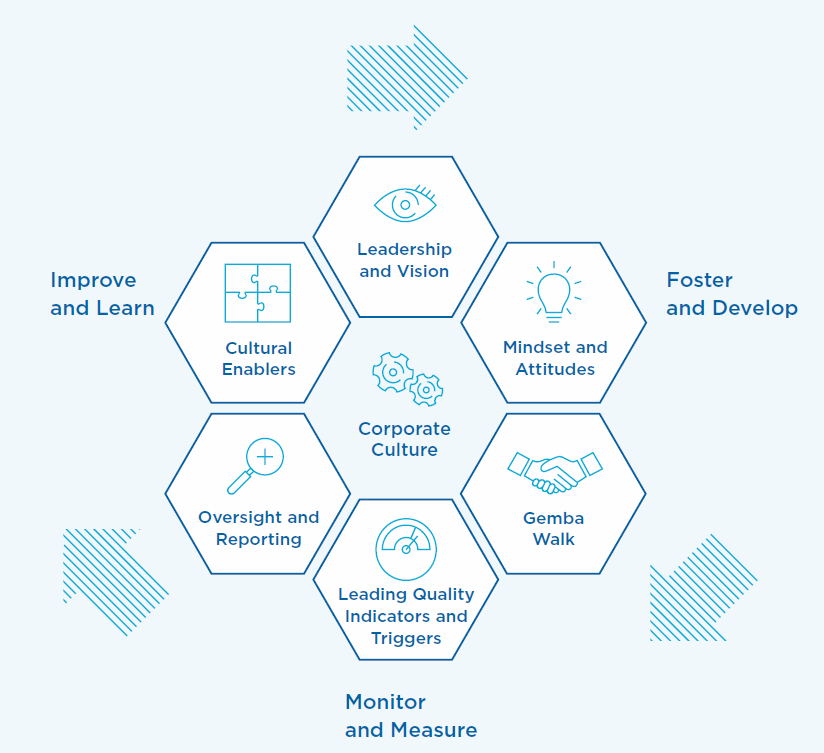
A “debilitated” safety department and too-thin maintenance crews are just two of the revelations surfaced by a new report on the MBTA’s safety culture.
Let’s cut right to the heart of the report’s recommendations:
“The panel makes six policy recommendations that are intended “to move the organization to a place where safety is a priority and is culturally integrated into every aspect of their mission.” They include establishing better safety performance indicators, identifying the areas where maintenance is being deferred, implementing stronger data collection, and strengthening the MBTA’s leadership team with “more seasoned” transit professionals.“
ICH Q12 “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management” was adopted by the ICH in Singapore, which means Q12 is now in Stage 5, Implementation. Implementation should be interesting as concepts like “established conditions” and “product lifecycle management” which sit at the core of Q12 are still open for interpretation as Q12 is implemented in specific regulatory markets.
This draft guidance is now in a review period by regulatory agencies. Which means no public comments, but it will be applied on a 6-month trial basis by PIC/S participating authorities, which include the US Food and Drug Administration and other regulators across Europe, Australia, Canada, South Africa, Turkey, Iran, Argentina and more.
This document is aligned to ICH Q10, and there should be few surprised in this. Given PIC/S concern that “ongoing continual improvement has probably not been realised to a meaningful extent. The PIC/S QRM Expert Circle, being well-placed to focus on the QRM concepts of the GMPs and of ICH Q10, is seeking to train GMP inspectors on what a good risk-based change management system can look like within the PQS, and how to assess the level of effectiveness of the PQS in this area” it is a good idea to start aligning to be ahead of the curve.
“Changes typically have an impact assessment performed within the change control system. However, an impact assessment is often not as comprehensive as a risk assessment for the proposed change.”
This is a critical thing that agencies have been discussing for years. There are a few key takeaways.
The difference between impact and risk is critical. Impact is best thought of as “What do I need to do to make the change.” Risk is “What could go wrong in making this change?” Impact focuses on assessing the impact of the proposed change on various things such as on current documentation, equipment cleaning processes, equipment qualification, process validation, training, etc. While these things are very important to assess, asking the question about what might go wrong is also important as it is an opportunity for companies to try to prevent problems that might be associated with the proposed change after its implementation.
This 8 page document is really focusing on the absence of clear links between risk assessments, proposed control strategies and the design of validation protocols.
The guidance is very concerned about appropriately classifying changes and using product data to drive decisions. While not specifying it in so many words, one of the first things that popped to my mind was around how we designate changes as like-for-like in the absence of supporting data. Changes that are assigned a like-for-like classification are often not risk-assessed, and are awarded limited oversight from a GMP perspective. These can sometimes result in major problems for companies, and one that I think people are way to quick to rush to.
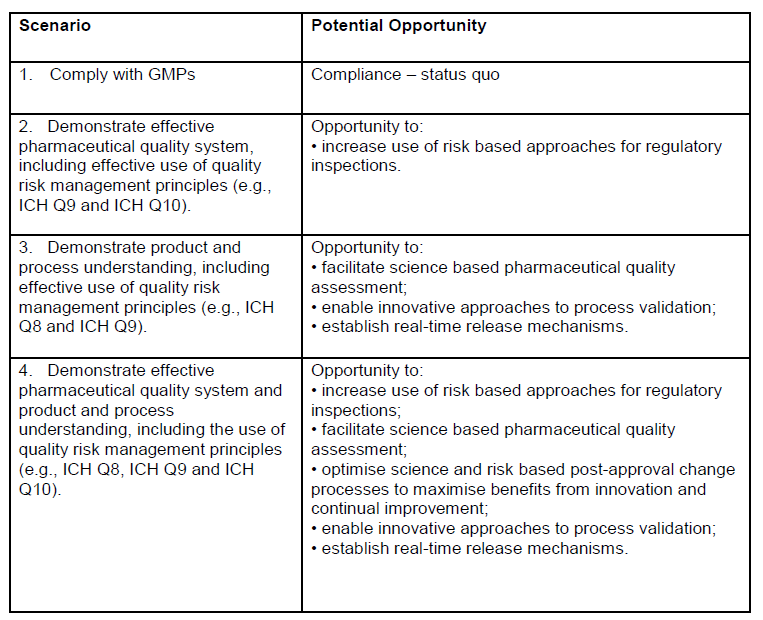
It is fascinating to look at appendix 1, which really lays out some critical goals of this draft guidance: better risk management, real time release, and innovative approaches to process validation. This is sort of the journey we are all on.
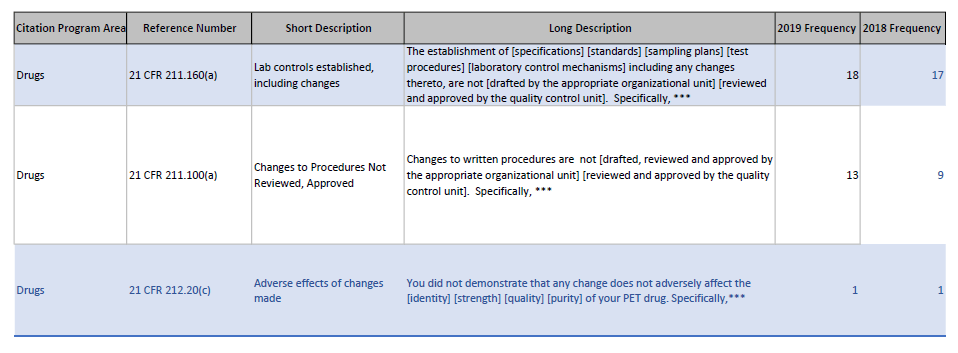
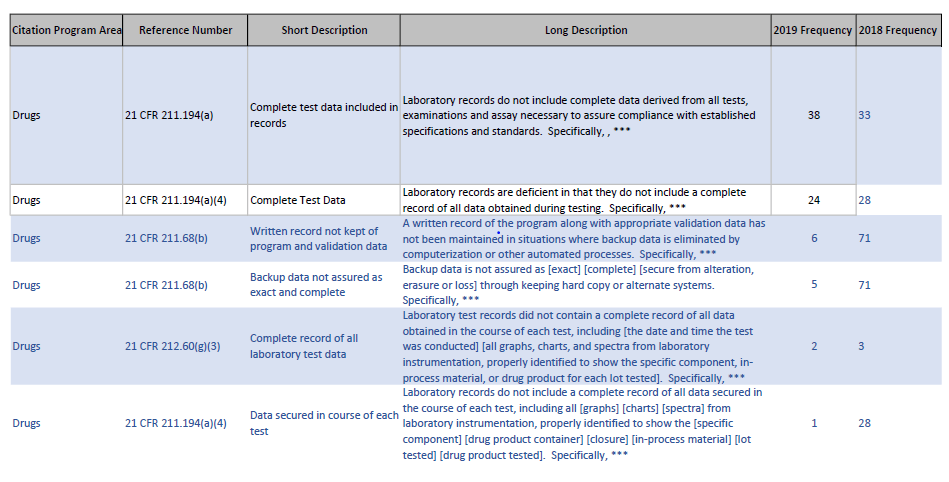
The FDA has posted the 2019 483 observations as an excel file. The FDA has made these files available every year since 2006 and I find them to be one of my favorite tools for evaluating regulatory trends.
So for example, looking at change related 483 I see:
2019 vs 2018 483 comparison for short description including “change”
Last November, officials from the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) met with industry representatives in London to discuss the various quality challenges that arise when the development of investigational products is accelerated .
The report was recently published, and can be found here.
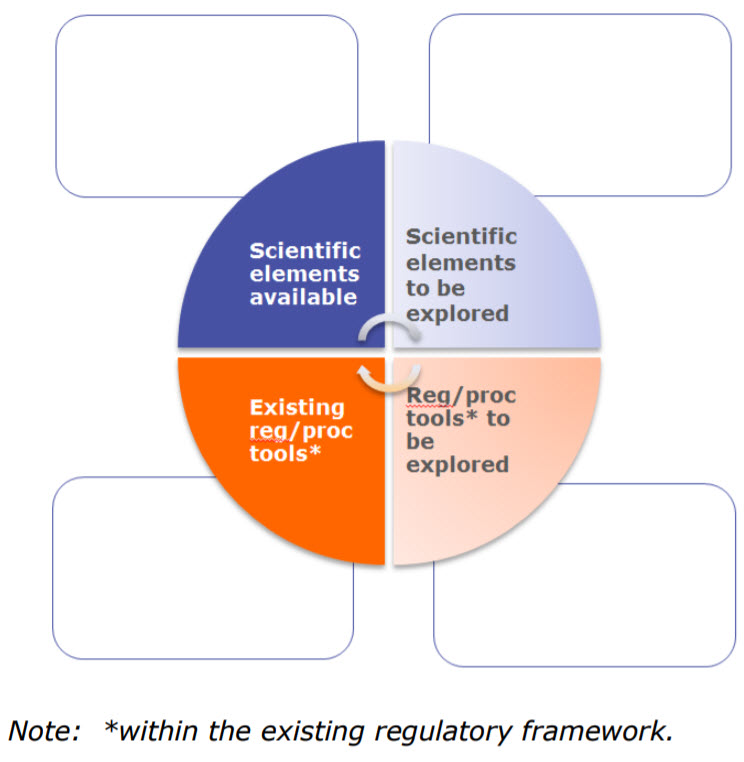
The workshop discussed process validation, control strategies, good manufacturing practice (GMP) compliance, comparability, stability and regulatory tools of early access approaches. Throughout they discussed two elements:
Scientific which includes technologies and scientific concepts or principles for development, manufacture and quality risk management, which may or not be present or implied in existing guidelines. Examples include concurrent validation, new modelling methodologies, new analytical techniques, etc.
Regulatory/procedural tools are described in the legal, regulatory framework and can be specific to PRIME (or Breakthrough Therapies) (e.g. kick-off meetings) or generally applicable [e.g. Post-approval change management protocols (PACMPs), recommendations, scientific advice (SA)].
I strongly recommend reading the report in it’s entirety.
Data Integrity definitely continues to be a theme, and I agree that we are seeing a growing trend around process validation. I also think root cause investigations was a theme of 2018 that we are going to be seeing a lot more of.