The flow chart is a simple, but important, graphic organizer. Placing the states or steps of an event or process into the correct sequence allows you to reach conclusions and make predictions.
However, its simplicity means we don’t always work to be consistent and can benefit from a little effort to ensure users are aligned.
I am a huge fan of including flow charts in all process and procedure documents.
Steps for Building a flow chart
Capture
Capture the events or steps of the process. Resist the urge to arrange them sequentially and concentrate on capturing the events/steps only.
Cull
If there are more than eight steps in a flow chart we start creating cognitive overload. If a process or procedure has more than eight steps you need to:
Ensure the steps are at the right level, sometimes we have substeps represented and we can cull that. Ensure they are all on the same level of process/procedure/task.
Decide we need to break the procedure into multiple documents. This is a great way to decide what work instructions are necessary.
Look for opportunity for process improvement.
Sequence the events and draw the flow chart
The focus now shifts to temporal relations. The correct sequential arrangements of steps or events helps to reach conclusions about past events and prepare for future events.
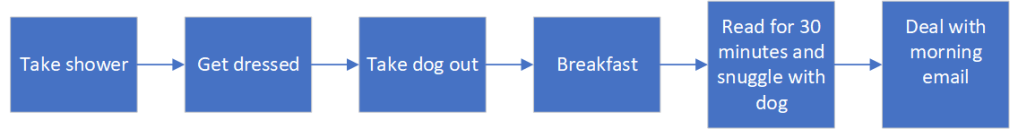
Example
I’m writing the procedure for my mornings, I capture the following:
Eat breakfast
Take shower
Take dog out
Get dressed
Decide on tea
Heat water
Drink tea
Read for 30 minutes
Deal with morning email
Snuggle with dog
Taking a look at the list I realize that not everything is on the same level of process/procedure/task and end up with a shorter list.
Breakfast
Take shower
Take dog out
Get dressed
Read for 30 minutes
Deal with morning email
Snuggle with dog
Notice how I combined all the tea stuff into a breakfast category. When brainstorming my list I put a lot of weight on tea, because it is important to me (yes I have been using tea as a training example since 2005, I just love tea).
I can then put them in sequence:
Flow Chart for my morning
When I was making things sequential I realized that two of my activities (read and dog snuggle) were concurrent, so I combined them as one step.
Risk can be associated with a number of different types of consequences, impacting different objectives. The types of consequences to be analyzed are decided when planning the assessment. The context statement is checked to ensure that the consequences to be analyzed align with the purpose of the assessment and the decisions to be made. This can be revisited during the assessment as more is learned.
Methods used in analyzing risks can be qualitative, semiquantitative, or quantitative. The decision here will be on the intended use, the availability of reliable data, and the decision-making needs of the organization. In ICH Q9 this is also the level of formality.
Risk
Is….
The combination of the probability of the occurrence of the harm
and the severity of that harm.
The effect of uncertainty on objectives
Often characterized by reference to the potential event and
consequences or combination of these
Often expressed in terms of a combination of the consequences of
an event (including in changes in circumstances) and the associated
likelihood of the occurrence
Qualitative assessments define consequence (or severity), likelihood, and level of risk by significance levels, such as “high,” “medium,” or “low.” They work best when supporting analysis that have a narrow application or are within another quality system, such as change control.
Qualitative
Below is a good way to break down consequences and likelihood for a less formal assessment.
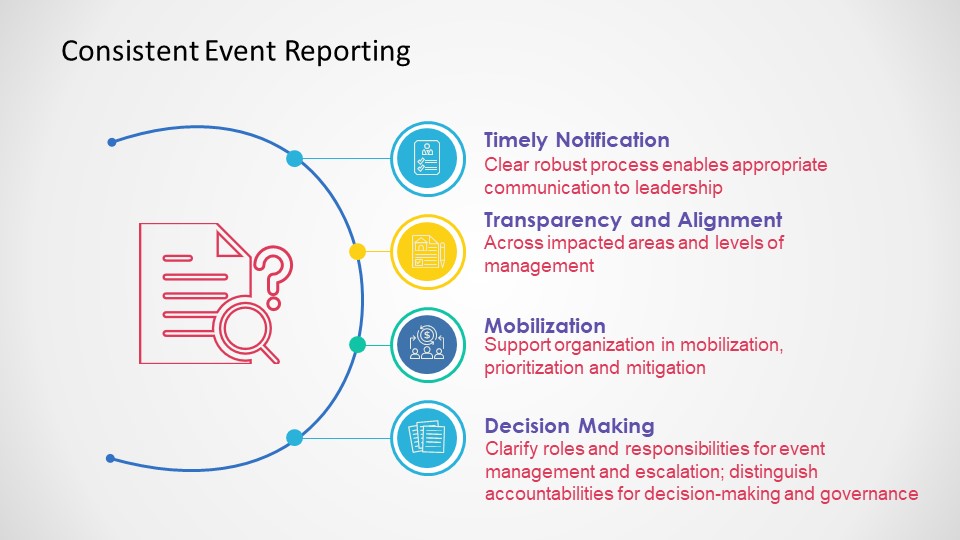
Event management systems need to have an escalation mechanism to ensure critical events are quickly elevated to a senior level to ensure organization-wide timely reactions.
Consistent Event Reporting
There are many reasons for a fast escalation.
Events that trigger reporting to Regulatory Agencies (e.g. Serious Breach, Urgent Safety Measures (UK), Field Alerts, Biological Product Deviation, Medical Device Report)
Events that require immediate action to prevent additional harm from across the organization
Events that require marshalling resources from large parts of the organization
•Reference GxP area for Impact
resulting from/linked to system error/failure
•Product Quality/ CMC events in
accordance with MRB criteria (or other events of similar scope of impact)
•Impact to study integrity
•Impact to subject’s safety, rights or
welfare
•Gaps in reporting/ collection of
potential AEs
•Impact to study integrity
•Impact to study integrity
•System design, testing, deployment,
upgrade, etc. event impacting GxP data integrity or regulatory compliance
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Potential Falsified or Counterfeit
Product
•Potential Fraud or Misconduct
•Potential Fraud or Misconduct
•Credible Risk of Product Shortage
•Quality event with patient safety
risk/gap
•GxP Data Breach
•Potential Product Recall
•Significant Quality Event Notified to
Regulatory Authority
•System error or failure with
significant GxP compliance impact
·Potential Critical Finding Resulting from
Regulatory Authority Inspection or Audit by External Body/Third Party
·Quality Event/Observation Classified
as Critical (Event or Internal Audit) Notification from Regulatory Authority
or other External Authority of Findings of Significant/Critical Quality
Deficiency (inspection or other than through inspection)
oe.g.; Refusal to File, Notification
of Inadequate Response to Inspection Findings (e.g.; Other Action Indicated
(FDA classification), Warning Letter
You can drill down to a lower, more practical level, like this
Escalation Criteria
Examples of Quality Events for Escalation
Potential to adversely affect
quality, safety, efficacy, performance or compliance of product (commercial
or clinical)
•Contamination (product, raw material,
equipment, micro; environmental)
•Product defect/deviation from process
parameters or specification (on file with agencies)
•Significant GMP deviations
•Incorrect/deficient labeling
•Product complaints (significant PC,
trends in PCs)
•OOS/OOT (e.g., stability)
Product counterfeiting, tampering, theft
•Product counterfeiting, tampering, theft reportable to Health
Authority (HA)
•Lost/stolen IMP
•Fraud or misconduct associated with counterfeiting, tampering,
theft
•Potential to impact product supply (e.g., removal, correction,
recall)
Product shortage likely to
disrupt patient care and/or reportable to HA
•Disruption of product supply due to
product quality events, natural disasters (business continuity disruption),
OOS impact, capacity constraints
Potential to cause patient harm associated with a product
quality event
•Urgent Safety Measure, Serious Breach, Significant Product
Compliant, Safety Signal that are determined associated with a product
quality event
Significant GMP
non-compliance/event
•Non-compliance or non-conformance
event with potential to impact product performance meeting specification,
safety efficacy or regulatory requirements
Regulatory Compliance Event
•Significant (critical, repeat) regulatory inspection findings,
lack of commitment adherence
•Notification of directed/for cause inspection
•Notification of HA correspondence indicating potential
regulatory action
An updated and expanded version of this is found here.
Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
Regulation/Guidance
States
ICH E-6 (R2) Section 4.5.1-4.5.4
4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion.
ICH E3, section 9.6
The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report
21CFR 312.53(vi) (a)
investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.”
21CFR 56.108(a)
IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects.
21 CFR 56.108(b)
“IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.”
45 CFR 46.103(b)(5)
Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB.
FDA Form-1572 (Section 9)
lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.”
A few key regulations and guidances (not meant to be a comprehensive list)
How Protocol Deviations are Implemented
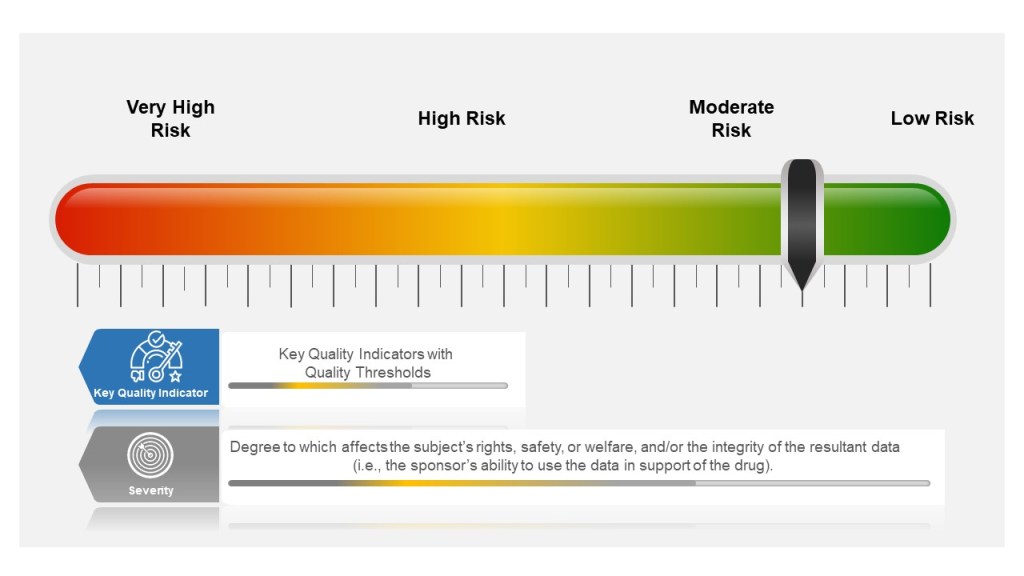
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
Events happen beyond the Protocol. These need to be managed appropriately as well.
The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Event Classification for Clinical Protocols and GCPs
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
Events Happen
Consistent Event Reporting is Critical
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are:
The FDA recently released a Form 483 it handed to Catalent Belgium following an inspection of its 265,000 square-foot facility in Brussels in October 2021. Catalent is a pretty sizable entity, so it is very valuable to see what we can learn from their observations.
Failure to adequately assess an unexplained discrepancy or deviation
“Standard Operating Procedure STB-QA-0010, Deviation Management, v21 classifies deviations as minor, major or critical based on the calculation of a risk priority number, with a HEPA filter failure within a Grade A environment often classified as minor. Specifically, Deviation 327567 (Date of occurrence 04 March 2021) was for a HEPA filter failure on the <redacted> fill line, with a breach at the HEPA filter frame.”
This one is more common than it should be. I’ve recently written about categorization and criticality of events. I want to stress the term potential when addressing impact in the classification of events.
Control barriers exist for a reason. You breach that control barrier in any way, you have the potential to impact product or environment. It is really easy for experienced SMEs to say “But this has never had any real impact before” and then downgrade the deviation classification. Before long it becomes the norm that HEPA filter failures are minor because they never have impact. And then one does. Then there are shortages or worse.
It is important to avoid that complacency and treat each and every control barrier failure to the same level of investigation based on their potentiality to impact.
The other problem here is failure to identify trends and deal with them. I can honestly say that the last thing I ever want anyone, especially an inspector, to write about something where I have quality oversight is a failure to investigate multiple control barrier events.
“Other GMP manufacturing areas have a similar elevated level of HEPA filter failures, with the root cause of the HEPA filter failures unknown. There is no CAPA in support of correction action. Your firm failed to ensure your investigations identify appropriate root causes and you failed to implement sustainable corrective action and preventive action (CAPA).“
Contamination Control function
Observation 2 and 3 are doozies, but there is probably a lack of expertise involved here. The site is using out-of-date and inadequate methods in their validation. Hire a strong contamination control expert and leverage them. Build expertise in the organization through a robust training program. Connect this to all relevant quality systems/processes.
Corrective Maintenance and Troubleshooting
“Equipment and facilities used in the manufacture of drug product are not adequately maintained or appropriately designed to facilitate operations for their intended use.“
This is starting to feel a lot like my upcoming presentation at the 2022 ISPE Aseptic Conference where I will be speaking on “Contamination Control, Risk and the Quality Management System”
“Contamination Control is a fairly wide term used to mean “getting microbiologists out of the lab” and involved in risk management and the quality management system. This presentation will evaluate best practices in building a contamination control strategy and ensuring its use throughout the quality system. Leveraging a House of Quality approach, participants will learn how to: Create targeted/ risk based measures of contamination avoidance; Implement Key performance indicators to assess status of contamination control; and ensure a defined strategy for deviation management (investigations), CAPA and change management.”