People who have gone through consent decrees and other regulatory challenges (and I know several individuals who have done so more than once) tend to joke that every year under a consent decree is equivalent to 10 years of experience anywhere else. There is something to this joke, as consent decrees represent unique opportunities for accelerated learning and expertise development that can fundamentally transform organizational capabilities. This phenomenon aligns with established scientific principles of learning under pressure and deliberate practice that your organization can harness to create sustainable, healthy development programs.
Understanding Consent Decrees and PAI/PLI as Learning Accelerators
A consent decree is a legal agreement between the FDA and a pharmaceutical company that typically emerges after serious violations of Good Manufacturing Practice (GMP) requirements. Similarly, Post-Approval Inspections (PAI) and Pre-License Inspections (PLI) create intense regulatory scrutiny that demands rapid organizational adaptation. These experiences share common characteristics that create powerful learning environments:
High-Stakes Context: Organizations face potential manufacturing shutdowns, product holds, and significant financial penalties, creating the psychological pressure that research shows can accelerate skill acquisition. Studies demonstrate that under high-pressure conditions, individuals with strong psychological resources—including self-efficacy and resilience—demonstrate faster initial skill acquisition compared to low-pressure scenarios.
Forced Focus on Systems Thinking: As outlined in the Excellence Triad framework, regulatory challenges force organizations to simultaneously pursue efficiency, effectiveness, and elegance in their quality systems. This integrated approach accelerates learning by requiring teams to think holistically about process interconnections rather than isolated procedures.
Third-Party Expert Integration: Consent decrees typically require independent oversight and expert guidance, creating what educational research identifies as optimal learning conditions with immediate feedback and mentorship. This aligns with deliberate practice principles that emphasize feedback, repetition, and progressive skill development.
The Science Behind Accelerated Learning Under Pressure
Recent neuroscience research reveals that fast learners demonstrate distinct brain activity patterns, particularly in visual processing regions and areas responsible for muscle movement planning and error correction. These findings suggest that high-pressure learning environments, when properly structured, can enhance neural plasticity and accelerate skill development.
The psychological mechanisms underlying accelerated learning under pressure operate through several pathways:
Stress Buffering: Individuals with high psychological resources can reframe stressful situations as challenges rather than threats, leading to improved performance outcomes. This aligns with the transactional model of stress and coping, where resource availability determines emotional responses to demanding situations.
Enhanced Attention and Focus: Pressure situations naturally eliminate distractions and force concentration on critical elements, creating conditions similar to what cognitive scientists call “desirable difficulties”. These challenging learning conditions promote deeper processing and better retention.
Evidence-Based Learning Strategies
Scientific research validates several strategies that can be leveraged during consent decree or PAI/PLI situations:
Retrieval Practice: Actively recalling information from memory strengthens neural pathways and improves long-term retention. This translates to regular assessment of procedure knowledge and systematic review of quality standards.
Spaced Practice: Distributing learning sessions over time rather than massing them together significantly improves retention. This principle supports the extended timelines typical of consent decree remediation efforts.
Interleaved Practice: Mixing different types of problems or skills during practice sessions enhances learning transfer and adaptability. This approach mirrors the multifaceted nature of regulatory compliance challenges.
Elaboration and Dual Coding: Connecting new information to existing knowledge and using both verbal and visual learning modes enhances comprehension and retention.
Creating Sustainable and Healthy Learning Programs
The Sustainability Imperative
Organizations must evolve beyond treating compliance as a checkbox exercise to embedding continuous readiness into their operational DNA. This transition requires sustainable learning practices that can be maintained long after regulatory pressure subsides.
- Cultural Integration: Sustainable learning requires embedding development activities into daily work rather than treating them as separate initiatives.
- Knowledge Transfer Systems: Sustainable programs must include systematic knowledge transfer mechanisms.
Healthy Learning Practices
Research emphasizes that accelerated learning must be balanced with psychological well-being to prevent burnout and ensure long-term effectiveness:
- Psychological Safety: Creating environments where team members can report near-misses and ask questions without fear promotes both learning and quality culture.
- Manageable Challenge Levels: Effective learning requires tasks that are challenging but not overwhelming. The deliberate practice framework emphasizes that practice must be designed for current skill levels while progressively increasing difficulty.
- Recovery and Reflection: Sustainable learning includes periods for consolidation and reflection. This prevents cognitive overload and allows for deeper processing of new information.
Program Management Framework
Successful management of regulatory learning initiatives requires dedicated program management infrastructure. Key components include:
- Governance Structure: Clear accountability lines with executive sponsorship and cross-functional representation ensure sustained commitment and resource allocation.
- Milestone Management: Breaking complex remediation into manageable phases with clear deliverables enables progress tracking and early success recognition. This approach aligns with research showing that perceived progress enhances motivation and engagement.
- Resource Allocation: Strategic management of resources tied to specific deliverables and outcomes optimizes learning transfer and cost-effectiveness.
Implementation Strategy
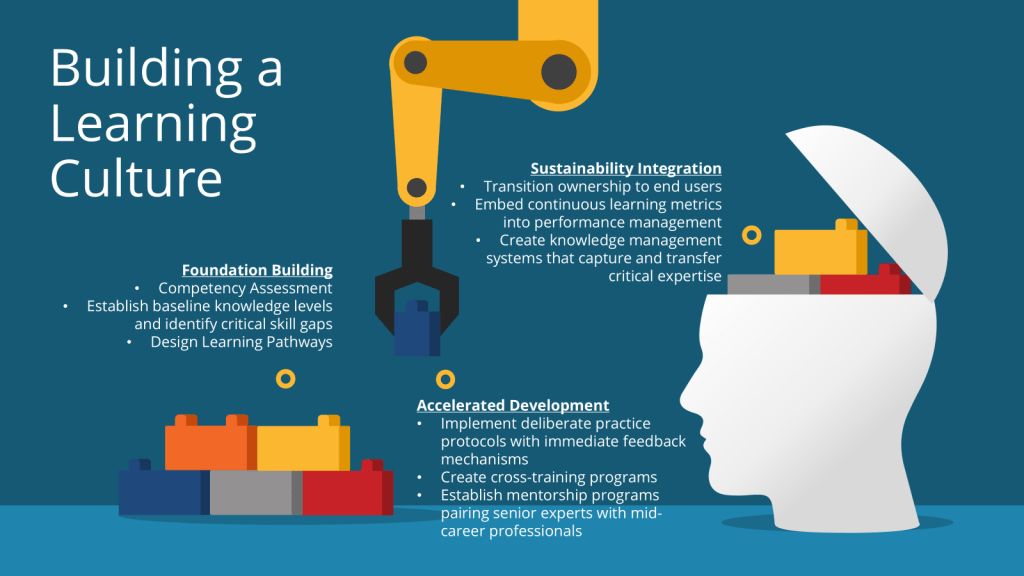
Phase 1: Foundation Building
- Conduct comprehensive competency assessments
- Establish baseline knowledge levels and identify critical skill gaps
- Design learning pathways that integrate regulatory requirements with operational excellence
Phase 2: Accelerated Development
- Implement deliberate practice protocols with immediate feedback mechanisms
- Create cross-training programs
- Establish mentorship programs pairing senior experts with mid-career professionals
Phase 3: Sustainability Integration
- Transition ownership of new systems and processes to end users
- Embed continuous learning metrics into performance management systems
- Create knowledge management systems that capture and transfer critical expertise
Measurement and Continuous Improvement
Leading Indicators:
- Competency assessment scores across critical skill areas
- Knowledge transfer effectiveness metrics
- Employee engagement and psychological safety measures
Lagging Indicators:
- Regulatory inspection outcomes
- System reliability and deviation rates
- Employee retention and career progression metrics
| Kirkpatrick Level | Category | Metric Type | Example | Purpose | Data Source |
|---|---|---|---|---|---|
| Level 1: Reaction | KPI | Leading | % Training Satisfaction Surveys Completed | Measures engagement and perceived relevance of GMP training | LMS (Learning Management System) |
| Level 1: Reaction | KRI | Leading | % Surveys with Negative Feedback (<70%) | Identifies risk of disengagement or poor training design | Survey Tools |
| Level 1: Reaction | KBI | Leading | Participation in Post-Training Feedback | Encourages proactive communication about training gaps | Attendance Logs |
| Level 2: Learning | KPI | Leading | Pre/Post-Training Quiz Pass Rate (≥90%) | Validates knowledge retention of GMP principles | Assessment Software |
| Level 2: Learning | KRI | Leading | % Trainees Requiring Remediation (>15%) | Predicts future compliance risks due to knowledge gaps | LMS Remediation Reports |
| Level 2: Learning | KBI | Lagging | Reduction in Knowledge Assessment Retakes | Validates long-term retention of GMP concepts | Training Records |
| Level 3: Behavior | KPI | Leading | Observed GMP Compliance Rate During Audits | Measures real-time application of training in daily workflows | Audit Checklists |
| Level 3: Behavior | KRI | Leading | Near-Miss Reports Linked to Training Gaps | Identifies emerging behavioral risks before incidents occur | QMS (Quality Management System) |
| Level 3: Behavior | KBI | Leading | Frequency of Peer-to-Peer Knowledge Sharing | Encourages a culture of continuous learning and collaboration | Meeting Logs |
| Level 4: Results | KPI | Lagging | % Reduction in Repeat Deviations Post-Training | Quantifies training’s impact on operational quality | Deviation Management Systems |
| Level 4: Results | KRI | Lagging | Audit Findings Related to Training Effectiveness | Reflects systemic training failures impacting compliance | Regulatory Audit Reports |
| Level 4: Results | KBI | Lagging | Employee Turnover | Assesses cultural impact of training on staff retention | HR Records |
| Level 2: Learning | KPI | Leading | Knowledge Retention Rate | % of critical knowledge retained after training or turnover | Post-training assessments, knowledge tests |
| Level 3: Behavior | KPI | Leading | Employee Participation Rate | % of staff engaging in knowledge-sharing activities | Participation logs, attendance records |
| Level 3: Behavior | KPI | Leading | Frequency of Knowledge Sharing Events | Number of formal/informal knowledge-sharing sessions in a period | Event calendars, meeting logs |
| Level 3: Behavior | KPI | Leading | Adoption Rate of Knowledge Tools | % of employees actively using knowledge systems | System usage analytics |
| Level 2: Learning | KPI | Leading | Search Effectiveness | Average time to retrieve information from knowledge systems | System logs, user surveys |
| Level 2: Learning | KPI | Lagging | Time to Proficiency | Average days for employees to reach full productivity | Onboarding records, manager assessments |
| Level 4: Results | KPI | Lagging | Reduction in Rework/Errors | % decrease in errors attributed to knowledge gaps | Deviation/error logs |
| Level 2: Learning | KPI | Lagging | Quality of Transferred Knowledge | Average rating of knowledge accuracy/usefulness | Peer reviews, user ratings |
| Level 3: Behavior | KPI | Lagging | Planned Activities Completed | % of scheduled knowledge transfer activities executed | Project management records |
| Level 4: Results | KPI | Lagging | Incidents from Knowledge Gaps | Number of operational errors/delays linked to insufficient knowledge | Incident reports, root cause analyses |
The Transformation Opportunity
Organizations that successfully leverage consent decrees and regulatory challenges as learning accelerators emerge with several competitive advantages:
- Enhanced Organizational Resilience: Teams develop adaptive capacity that serves them well beyond the initial regulatory challenge. This creates “always-ready” systems, where quality becomes a strategic asset rather than a cost center.
- Accelerated Digital Maturation: Regulatory pressure often catalyzes adoption of data-centric approaches that improve efficiency and effectiveness.
- Cultural Evolution: The shared experience of overcoming regulatory challenges can strengthen team cohesion and commitment to quality excellence. This cultural transformation often outlasts the specific regulatory requirements that initiated it.
Conclusion
Consent decrees, PAI, and PLI experiences, while challenging, represent unique opportunities for accelerated organizational learning and expertise development. By applying evidence-based learning strategies within a structured program management framework, organizations can transform regulatory pressure into sustainable competitive advantage.
The key lies in recognizing these experiences not as temporary compliance exercises but as catalysts for fundamental capability building. Organizations that embrace this perspective, supported by scientific principles of accelerated learning and sustainable development practices, emerge stronger, more capable, and better positioned for long-term success in increasingly complex regulatory environments.
Success requires balancing the urgency of regulatory compliance with the patience needed for deep, sustainable learning. When properly managed, these experiences create organizational transformation that extends far beyond the immediate regulatory requirements, establishing foundations for continuous excellence and innovation. Smart organizations can utilzie the same principles to drive improvement.
Some Further Reading
| Topic | Source/Study | Key Finding/Contribution |
| Accelerated Learning Techniques | https://soeonline.american.edu/blog/accelerated-learning-techniques/ https://vanguardgiftedacademy.org/latest-news/the-science-behind-accelerated-learning-principles | Evidence-based methods (retrieval, spacing, etc.) |
| Stress & Learning | https://pmc.ncbi.nlm.nih.gov/articles/PMC5201132/ https://www.nature.com/articles/npjscilearn201611 | Moderate stress can help, chronic stress harms |
| Deliberate Practice | https://graphics8.nytimes.com/images/blogs/freakonomics/pdf/DeliberatePractice(PsychologicalReview).pdf | Structured, feedback-rich practice builds expertise |
| Psychological Safety | https://www.nature.com/articles/s41599-024-04037-7 | Essential for team learning and innovation |
| Organizational Learning | https://journals.scholarpublishing.org/index.php/ASSRJ/article/download/4085/2492/10693 https://www.elibrary.imf.org/display/book/9781475546675/ch007.xml | Regulatory pressure can drive learning if managed |