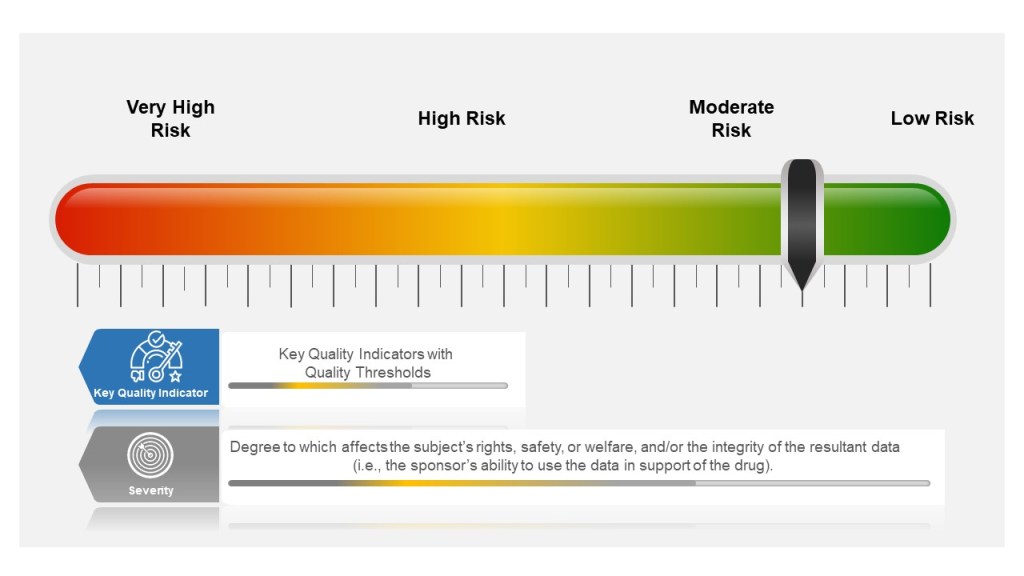
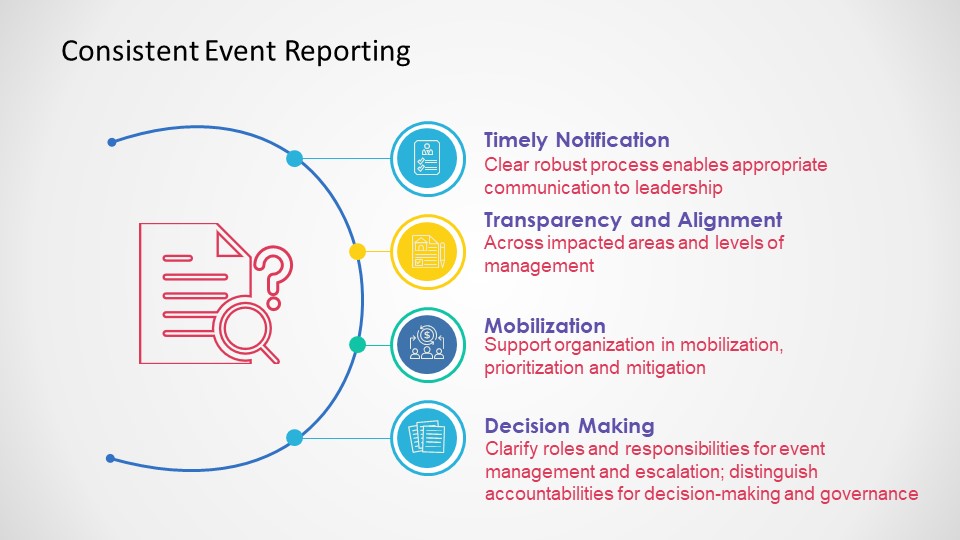
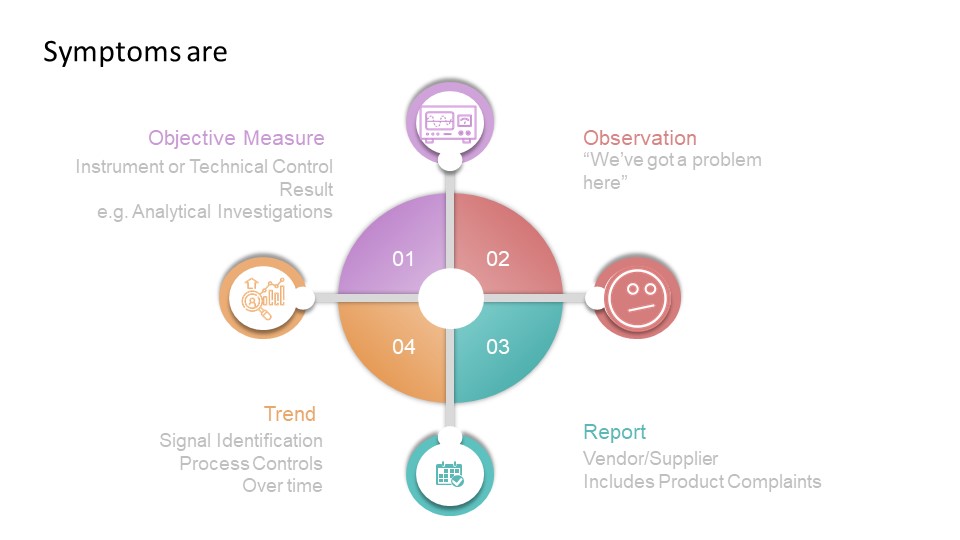
A list of Regulatory documents that apply to contamination control.
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 3: Premises and Equipment, (2014)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 5: Production, (2014)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Part II: Basic Requirements for Active Substances used as Starting Materials, (2014)
- European Union, Guidelines of 19 March 2015 on the formalized risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use, Official Journal of the European Union, (2015/C 95/02), (2015)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use, (2018)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 3 Manufacture of Radiopharmaceuticals, (2008)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 14 Manufacture of Medicinal Products Derived from Human Blood or Plasma, (2011)
- European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products, (2017)
- European Union, Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, Official Journal of the European Union, (2013/C 343/01), (2013),
- European Union, Guidelines of 19 March 2015 on principles of Good Distribution Practice of active substances for medicinal products for human use, Official Journal of the European Union, (2015/C 95/01), (2015)
- EMA Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (20 November 2014)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, subpart C = Building and Facilities, sec. 211.42 Design and construction features (b), (c)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart F – Production and Process Controls, sec. 211.113 Control of microbial contamination (a), (b)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart B – Organization and Personnel, sec.211.28 Personnel responsibilities (a)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control of Components and Drug Product Containers and Closures, sec. 211.80 General requirements. (b)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control of Components and Drug Product Containers and Closures, sec. 211.84 Testing and approval or rejection of components, drug product containers, and closures (d)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart D – Equipment, sec. 211.67 Equipment cleaning and maintenance (a)
- U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart C – Buildings and Facilities, sec. 211.56 Sanitation (c)
- U.S. Food & Drug Administration, Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, (2004)
- U.S. Food & Drug Administration, Guidance for Industry – Good Manufacturing Practice Considerations for Responding to COVID-19 Infection in Employees in Drug and Biological Products Manufacturing, (2020)
- U.S. Food & Drug Administration, Guidance for Industry – Guidance for Industry Non-Penicillin Beta-Lactam Drugs: A CGMP Framework for Preventing Cross Contamination, (2013)
- U.S. Food & Drug Administrationn, Guidance for Industry Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act, Draft Guidance. https://www.fda.gov/media/88905/download (accessed Mar 6, 2022)
- Pharmaceutical Inspection Co-operation Scheme gmp guide, 2nd targeted consultation document on revision of annex 1
- Pharmaceutical Inspection Co-operation Schemepharmaceutical inspection co-operation scheme gmp guide, ps inf 25 2019 (rev. 1) draft, manufacture of advanced therapy medicinal products for human use
- Pharmaceutical Inspection Co-operation Scheme gmp guide, ps inf 26 2019 (rev. 1) draft, manufacture of biological medicinal substances and products for human use
- Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part i), guide to good manufacturing practice for medicinal products part i
- Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part ii), guide to good manufacturing practice for medicinal products part ii
- Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (annexes), guide to good manufacturing practice for medicinal products annexes
- World Health Organisation, good manufacturing practices for pharmaceutical products: main principles, annex 2, who technical report series 986, 2014,
- World Health Organisation, who good manufacturing practices for active pharmaceutical ingredients (bulk drug substances), annex 2, who technical report series 957, 2010
- World Health Organisation, points to consider for manufacturers and inspectors: environmental aspects of manufacturing for the prevention of antimicrobial resistance annex 6, who technical report series 1025, 2020
- World Health Organisation, WHO good manufacturing practices for sterile pharmaceutical products, annex 6, who technical report series 961, 2011
- World Health Organisation, WHO good manufacturing practices for biological products, annex 3, who technical report series 996, 2016
- World Health Organisation, WHO good manufacturing practices for the manufacture of investigational pharmaceutical products for clinical trials in humans, annex 7, who technical report series 863, 1996
- World Health Organisation, WHO good manufacturing practices for radiopharmaceutical products annex 2, who technical report series 1025, 2020
- World Health Organisation, WHO GMP for Pharmaceutical Products containing Hazardous Substances, TRS 957, Annex-3 (2010)
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, Quality Risk Management, Q8 (R2), Pharmaceutical Development, August 2009. https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf (Accessed Mar 06, 2022)
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, Quality Risk Management Q9, November. https://database.ich.org/sites/default/files/Q9%20Guideline.pdf (accessed Mar 06, 2022).
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, pharmaceutical quality system Q10. https://database.ich.org/sites/default/files/Q10%20Guideline.pdf (accessed Mar 06, 2022).