I often get asked why I moved from a broader senior role in Quality Management to a particular but deep role in Quality Engineering and Validation. There are many answers, but the biggest is that validation is poised for some exciting shifts due to navigating a complex validation landscape characterized by rapid technological advancements, evolving regulatory standards, and the development of novel therapies. Addressing these challenges requires innovation, collaboration, and a proactive approach to risk management and data integration. Topics near and dear to me.
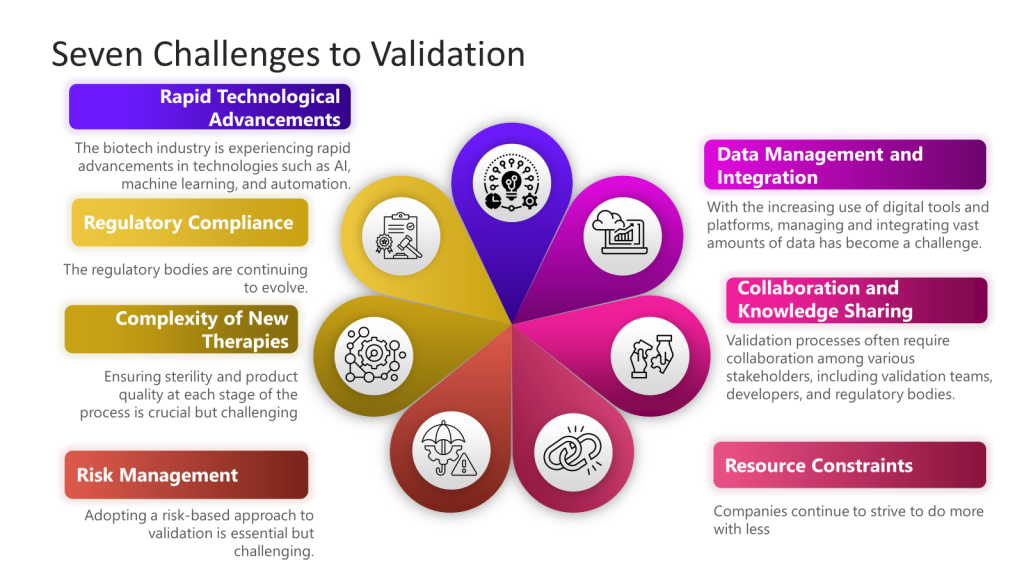
Today’s Challenges in Biotech Validation
1. Rapid Technological Advancements
The biotech industry is experiencing rapid technological advancements such as AI, machine learning, and automation. Integrating these technologies into validation processes can be challenging due to the need for new validation frameworks and methodologies.
2. Regulatory Compliance
Maintaining compliance with evolving regulatory standards is a significant challenge. Regulatory bodies like the FDA continuously update guidelines for technological advancements.
3. Complexity of New Therapies
Developing novel therapies, such as cell and gene therapies, introduces additional complexity to the validation process. These therapies often require redesigned facilities and equipment to accommodate their sensitive and sterile nature. Ensuring sterility and product quality at each process stage is crucial but challenging.
4. Data Management and Integration
Managing and integrating vast amounts of data has become challenging with the increasing use of digital tools and platforms. Effective data management is essential for predictive modeling and risk management in validation processes. Organizations must adopt robust data analytics and machine learning tools to handle this data efficiently.
Validation processes often require collaboration among various stakeholders, including validation teams, developers, and regulatory bodies. Ensuring real-time communication and data sharing can be challenging but is essential for streamlining validation efforts and aligning goals.
6. Resource Constraints
Smaller biotech companies, in particular, face resource constraints regarding funding, personnel, and expertise. These constraints can hinder their ability to implement advanced validation techniques and maintain compliance with regulatory standards.
Adopting a risk-based approach to validation is essential but challenging. Companies must identify and mitigate risks throughout the product lifecycle, which requires a thorough understanding of potential risks and effective risk management strategies.
Let’s Avoid the Term Validation 4.0
Let’s avoid the 4.0 term. We are constantly evolving, and adding a current ‘buzziness’ to it does no one any favors. We are shifting from traditional, paper-heavy validation methods to a more dynamic, data-driven, and digitalized process. Yes, we are leveraging modern technologies such as automation, data analytics, artificial intelligence (AI), and the Internet of Things (IoT) to enhance validation processes’ efficiency, flexibility, and reliability. But we don’t need buzziness, we just need to give it some thought, experiment, and refine.
I recently got asked what a medical device component manufacturer’s validation requirements are. Here is my answer.
Component manufacturers play a crucial role in the medical device industry by producing various parts and components for proper functioning and assembly. Here are some key expectations and responsibilities of component manufacturers in the medical device sector:
Quality and Precision Manufacturing: Medical device components often require high precision, accuracy, and quality to ensure patient safety and device efficacy. To meet these demanding standards, component manufacturers must adhere to stringent quality control measures, utilize advanced manufacturing techniques, and maintain strict tolerances.
Regulatory Compliance: The medical device industry is heavily regulated, and component manufacturers must comply with relevant regulations and standards set by governing bodies like the FDA, ISO, and others. This includes maintaining proper documentation, implementing quality management systems, and ensuring traceability of materials and processes.
Material Selection and Biocompatibility: Many medical device components come into direct contact with the human body or bodily fluids. Consequently, component manufacturers must carefully select biocompatible, non-toxic, and suitable materials for the intended application. They must also ensure proper sterilization and packaging to maintain sterility.
Design and Engineering Support: Some component manufacturers offer design and engineering services in addition to manufacturing to assist medical device companies in developing new components or optimizing existing ones. This collaboration helps ensure that components meet specific performance, functional, and regulatory requirements.
Supply Chain Management: Component manufacturers must have robust supply chain management systems to ensure the timely delivery of components to medical device manufacturers. This includes maintaining adequate inventory levels, managing logistics, and minimizing disruptions in the supply chain.
Yes, component manufacturers in the medical device industry are expected to validate their manufacturing processes to ensure the components they produce meet specified requirements and perform as intended.
Regulatory bodies like the FDA require that components critical to the safety and performance of medical devices be produced through validated processes. This helps ensure that components consistently meet quality standards.
Component manufacturers must perform Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) on their manufacturing equipment and processes.
Validation requirements apply to finished components and raw materials, sub-components received from suppliers, and any processes involved in producing the component. Traceability of validation activities throughout the supply chain is essential.
The level of validation required depends on the component’s criticality and risk to the final medical device. More stringent validation is expected for higher-risk components that directly contact the patient or are essential for device safety and efficacy.
The component manufacturer must maintain validation documentation such as protocols, test reports, and traceability matrices and provide it to the medical device company upon request for review and auditing purposes.
There have been a lot of changes in the way pharma thinks of analytical lifecycles in the last few years. With changes in technology, new product modalities, ICH Q2(R2) and ICH Q14 being released in November 2023, and USP <1220> in 2022, it is fair to say we are all catching up with our analytical lifecycle programs.
Let’s discuss what I think are the four pivotal documents that provide direction.
ICH Q2(R2) and ICH Q14
ICH Q2(R2) and ICH Q14 are complementary guidelines that provide a comprehensive framework for the development, validation, and lifecycle management of analytical procedures used in the pharmaceutical industry.
ICH Q14 describes the scientific principles and risk-based approaches for developing and maintaining suitable analytical procedures throughout their lifecycle. It outlines the key elements and considerations for analytical procedure development, including:
Defining an Analytical Target Profile (ATP)
Knowledge management and risk assessment
Evaluating robustness and parameter ranges
Establishing an Analytical Procedure Control Strategy
Lifecycle management and post-approval changes
Multivariate analytical procedures
Real-time release testing
On the other hand, ICH Q2(R2) provides specific guidance on validating analytical procedures to demonstrate their suitability for the intended use. It covers various validation tests, methodologies, and evaluation criteria, such as:
Specificity/selectivity
Working range
Accuracy and precision
Robustness
Stability-indicating properties
Multivariate analytical procedures
In summary, ICH Q14 establishes the overarching principles and approaches for analytical procedure development. At the same time, ICH Q2(R2) focuses on the validation aspects to ensure the analytical procedures are fit for purpose and meet quality requirements throughout their lifecycle. The two guidelines are intended to be applied together, with ICH Q14 providing the framework for development and ICH Q2(R2) specifying the validation requirements.
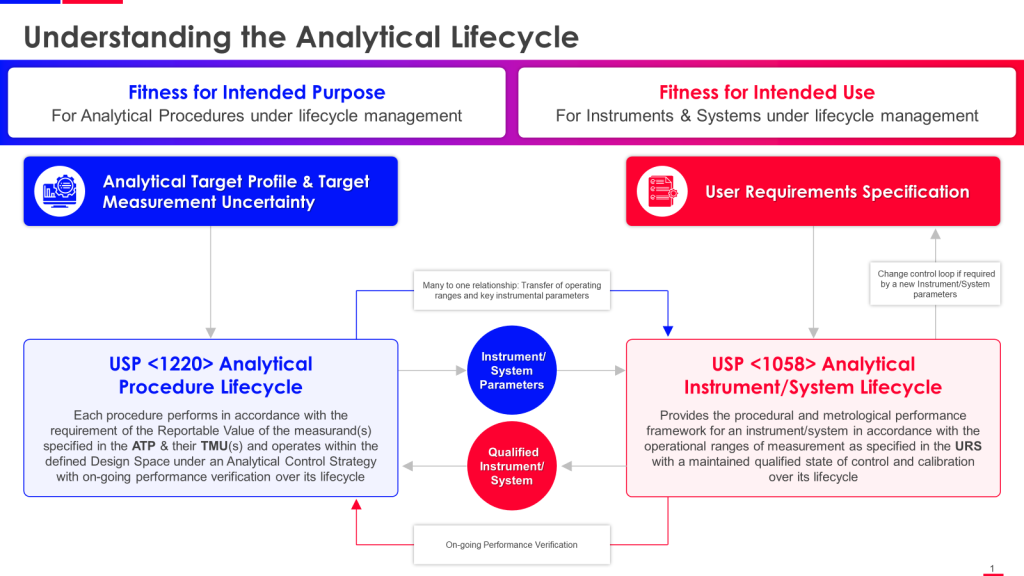
USP <1220> Analytical Procedure Lifecycle and USP <1058> Analytical Instrument Qualification are closely connected and complementary guidelines that provide a comprehensive framework for ensuring data integrity and quality in analytical procedures throughout their lifecycle.
The key connections between USP <1220> and USP <1058> are:
USP <1220> establishes the principles and requirements for managing the entire lifecycle of analytical procedures, from procedure design and development to retirement. It emphasizes the importance of defining an Analytical Target Profile (ATP) and implementing an Analytical Procedure Control Strategy.
USP <1058> focuses explicitly on the qualification of analytical instruments that execute analytical procedures. It outlines the requirements for ensuring instruments are suitable for their intended use through proper qualification (Design, Installation, Operational, and Performance Qualification).
The instrument qualification activities described in USP <1058> are critical to the overall Analytical Procedure Control Strategy outlined in USP <1220>. Proper instrument qualification as per <1058> helps ensure the quality and integrity of data generated by analytical procedures throughout their lifecycle.
Both guidelines stress the importance of defining user requirements (ATP in <1220> and User Requirements Specification in <1058>) as the basis for procedure development and instrument qualification activities.
USP <1220> requires ongoing monitoring and periodic requalification of analytical procedures, which includes re-evaluating the suitability of the analytical instruments used, as described in the Performance Qualification section of <1058>.
USP <1220> provides the overarching framework for holistically managing analytical procedures. USP <1058> focuses on ensuring the instruments used to execute those procedures are properly qualified and suitable for their intended use. The two guidelines work together to maintain data integrity and quality across the entire analytical lifecycle.
Complementary Approaches
USP <1220> Analytical Procedure Lifecycle is closely related to and complements the ICH Q2(R2) and ICH Q14 guidelines.
USP <1220> aligns with the principles outlined in ICH Q14 for managing the entire lifecycle of analytical procedures, from design and development to retirement. Both emphasize defining an Analytical Target Profile and implementing an Analytical Procedure Control Strategy.
The validation activities described in ICH Q2(R2), such as evaluating specificity, accuracy, precision, and robustness, are critical components of the Analytical Procedure Control Strategy required by USP <1220>.
USP <1220> requires ongoing monitoring and periodic requalification of analytical procedures, which aligns with the lifecycle management approach promoted in ICH Q14 and the validation during the lifecycle section in Q2(R2).
All these guidelines stress the importance of knowledge management, risk management, and a science/risk-based approach throughout the analytical procedure lifecycle.
The instrument qualification requirements outlined in USP <1058> are an integral part of the overall Analytical Procedure Control Strategy described in USP <1220>, ensuring instruments are suitable as per ICH Q2(R2) validation principles.
In essence, USP <1220> provides a comprehensive framework for analytical procedure lifecycle management that incorporates and operationalizes the scientific principles and validation activities detailed in the ICH Q14 and Q2(R2) guidelines, while USP <1058> provides the roadmap for instrument qualification. These four documents establish harmonized best practices for analytical procedures from development through retirement.
Commissioning, qualification, and validation are three distinct but interrelated processes in the pharmaceutical and biotechnology industries that ensure facilities, equipment, systems, and processes meet regulatory requirements and produce products of the desired quality. Here are the key differences:
Commissioning
Commissioning is a systematic process of ensuring that equipment, systems, and facilities are designed, installed, and functioning according to operational and engineering requirements.
It involves design reviews, installation verification, functional testing, and handover to operations.
Commissioning primarily focuses on satisfying engineering requirements and does not have direct regulatory requirements.
Qualification
Qualification is a regulated and documented process that demonstrates that equipment, systems, and facilities are installed correctly and operate as intended for their specific use.
It applies only to equipment, systems, and utilities that directly or indirectly impact product quality and patient safety.
Qualification activities include Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ).
Qualification is focused on by regulatory authorities like the FDA and EMA to ensure compliance.
Validation
Validation is a broader concept establishing documented evidence that a process consistently produces a product that meets its predetermined specifications and quality attributes.
It encompasses the entire process lifecycle, including process design, qualification of equipment/systems, and continued process verification.
Validation ensures that the equipment and systems are qualified and the entire process is controlled to produce the desired final product.
In summary, commissioning verifies engineering requirements, qualification demonstrates suitability for intended use, and validation provides a high degree of assurance that the process will consistently produce a quality product. These activities are interconnected, with commissioning often leveraged during qualification and qualification being a subset of the overall validation process.
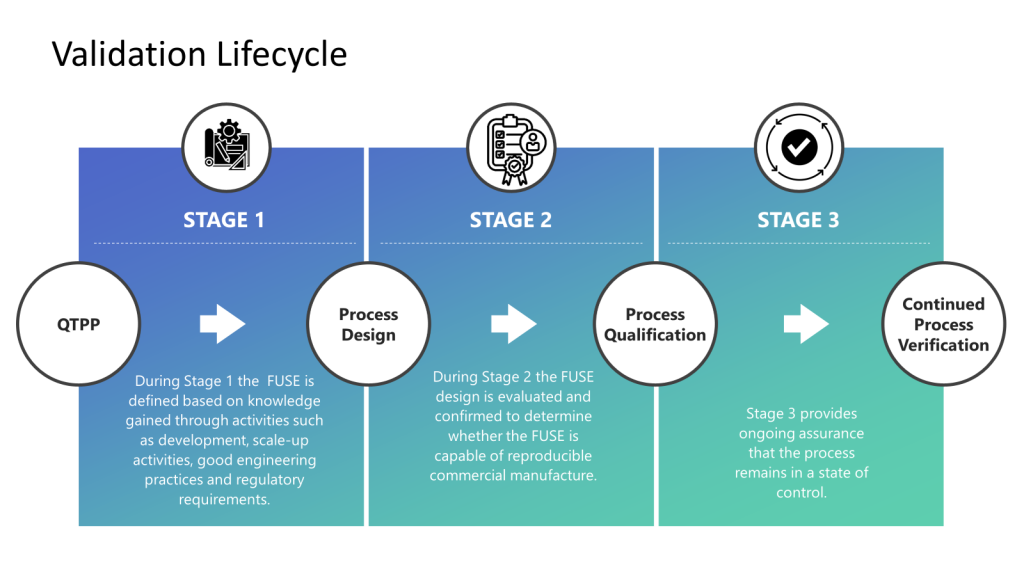
FDA’s Framework for Process Validation
The FDA’s Process Validation Guidance is a core document outlining a lifecycle approach with outlines a lifecycle approach with three main stages:
Stage 1: Process Design
Establish a process design based on knowledge gained through development and scale-up activities.
Identify critical quality attributes (CQAs) and critical process parameters (CPPs) using risk assessment and multivariate studies like Design of Experiments (DoE).
Develop a control strategy to ensure CQAs are met.
Stage 2: Process Qualification
Evaluate the process design through facility, utility, and equipment qualification.
Conduct performance qualification (PQ) by running production batches to confirm the process design has reproducible commercial manufacturing.
Establish scientific evidence that the process meets all defined requirements and product specifications.
Stage 3: Continued Process Verification
Maintain the validated status and monitor performance to ensure a state of control.
Conduct product quality reviews periodically to evaluate process performance.
The guidance emphasizes using a science and risk-based approach throughout the lifecycle, leveraging process understanding and knowledge gained from development through commercial production. Effective process validation requires good planning, documented evidence, and a robust quality system.
The User Requirements are a foundational document identifying the system’s product and process requirements. These product quality-related user requirements are based on product knowledge (CQAs), process knowledge (CPPs), regulatory requirements, and organization/site quality requirements. Writing a good user requirement for quality requirements involves several critical steps to ensure clarity, specificity, and effectiveness.
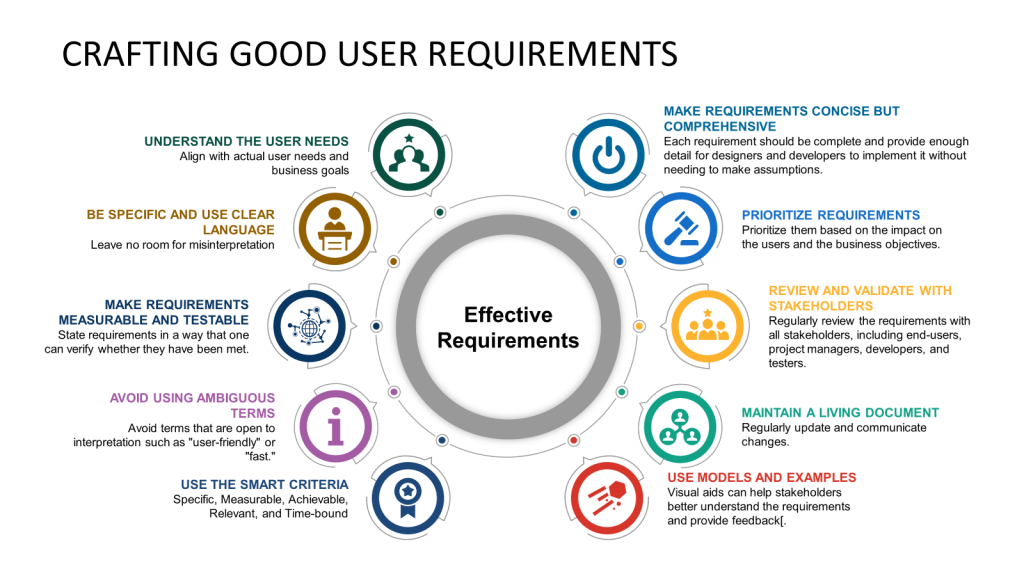
Understand the User Needs
Start by thoroughly understanding the user’s needs. This involves engaging with the end users or stakeholders to gather insights about their expectations, pain points, and the context in which the system will be used. This foundational step ensures that the requirements you develop are aligned with actual user needs and business goals.
Be Specific and Use Clear Language
Requirements should be specific and clearly stated to avoid ambiguity. Use simple, direct language and avoid technical jargon unless it is widely understood by all stakeholders. Define all terms and ensure that each requirement is phrased in a way that leaves no room for misinterpretation.
Make Requirements Measurable and Testable
Each requirement should be measurable and testable. This means stating requirements so one can verify whether they have been met. For example, instead of saying, “The system should load fast,” specify, “The system should load within 3 seconds when the number of simultaneous users is less than 10,000”.
Avoid Using Ambiguous Terms
Avoid terms open to interpretation, such as “user-friendly” or “fast.” If such terms are necessary, clearly define what they specifically mean in the context of your project. For instance, “user-friendly” is “the user can complete the desired task with no more than three clicks”.
Use the SMART Criteria
Employ the SMART criteria to ensure that each requirement is Specific, Measurable, Achievable, Relevant, and Time-bound. This approach helps set clear expectations and facilitates easier validation and verification of the requirements.
Make Requirements Concise but Comprehensive
While keeping each requirement concise and to the point is important, ensure all necessary details are included. Each requirement should be complete and provide enough detail for designers and developers to implement without making assumptions.
Prioritize Requirements
Not all requirements are equally important. Prioritize them based on their impact on the users and the business objectives. This helps manage the project scope and focuses on delivering maximum value.
It is good to categorize the user requirements here, such as:
Quality
Business
Health, Safety, and Environmental (HSE)
Review and Validate with Stakeholders
Review the requirements regularly with all stakeholders, including end-users, project managers, developers, and testers. This collaborative approach helps identify gaps or misunderstandings early in the project lifecycle.
Maintain a Living Document
Requirements might evolve as new information emerges or business needs change. Maintain your requirements document as a living document, regularly update it, and communicate changes to all stakeholders.
Use Models and Examples
Where applicable, use diagrams, mock-ups, or prototypes to complement the written requirements. Visual aids can help stakeholders better understand the requirements and provide feedback.
When writing user requirements for quality requirements, it’s crucial to avoid common pitfalls that can lead to misunderstandings, scope creep, and, ultimately, a product that does not meet the needs of the users or stakeholders. Here are some of the most common mistakes to avoid:
Ambiguity and Lack of Clarity
One of the most frequent errors in writing requirements is ambiguity. Requirements should be clear and concise, with no room for interpretation. Using vague terms like “user-friendly” or “fast” without specific definitions can lead to unmet expectations because different people may interpret these terms differently.
Incomplete Requirements
Another common issue is incomplete requirements that do not capture all necessary details or scenarios. This can result in features that do not fully address the users’ needs or require costly revisions later in development.
Overlooking Non-Functional Requirements
Focusing solely on what the system should do (functional requirements) without considering how it should perform (non-functional requirements), such as performance, security, and usability, can jeopardize the system’s effectiveness and user satisfaction.
Failure to Involve Stakeholders
Not involving all relevant stakeholders in the requirements gathering and validation process can lead to missing critical insights or requirements important to different user groups. This often results in a product that does not fully meet the needs of all its users.
Scope Creep
Without a clear definition of scope, projects can suffer from scope creep, where additional features and requirements are added without proper review, leading to delays and budget overruns. It’s important to have a well-defined project scope and a change management process in place.
Not Prioritizing Requirements
Not all requirements are equally important. Failing to prioritize requirements can misallocate resources and efforts on less critical features. Using prioritization techniques like MoSCoW (Must have, Should have, Could have, Won’t have this time) can help manage and focus efforts on what truly matters.
Lack of Validation and Verification
Skipping the validation (ensuring the product meets the intended use and needs of the stakeholders) and verification (ensuring the product meets the specified requirements) processes can lead to a final product not aligned with user needs and expectations.
Poor Documentation and Traceability
Inadequate documentation and lack of traceability can lead to confusion and errors during development. Maintaining detailed documentation and traceability from requirements through to implementation is crucial to ensure consistency and completeness.
Ignoring the Importance of Clear Communication
Effective communication is essential throughout the requirements process. Miscommunication can lead to incorrect or misunderstood requirements being developed. Regular, clear communication and documentation updates are necessary to keep all stakeholders aligned.
Not Considering the Testing of Requirements
Considering how requirements will be tested during the definition phase is important. This consideration helps ensure that requirements are testable and that the final product will meet them. Planning for testing early can also highlight any potential issues with clarity or feasibility of requirements.