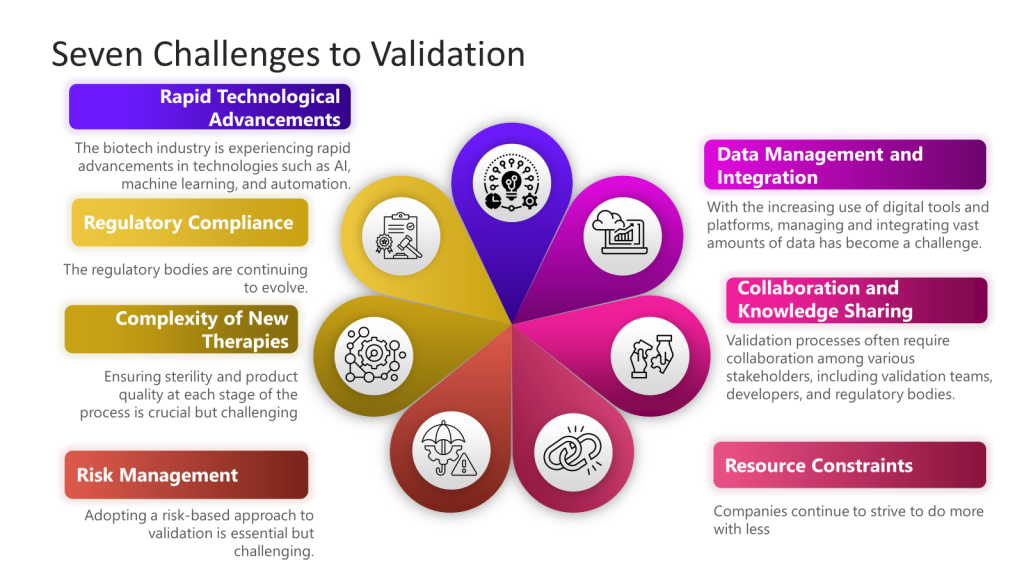
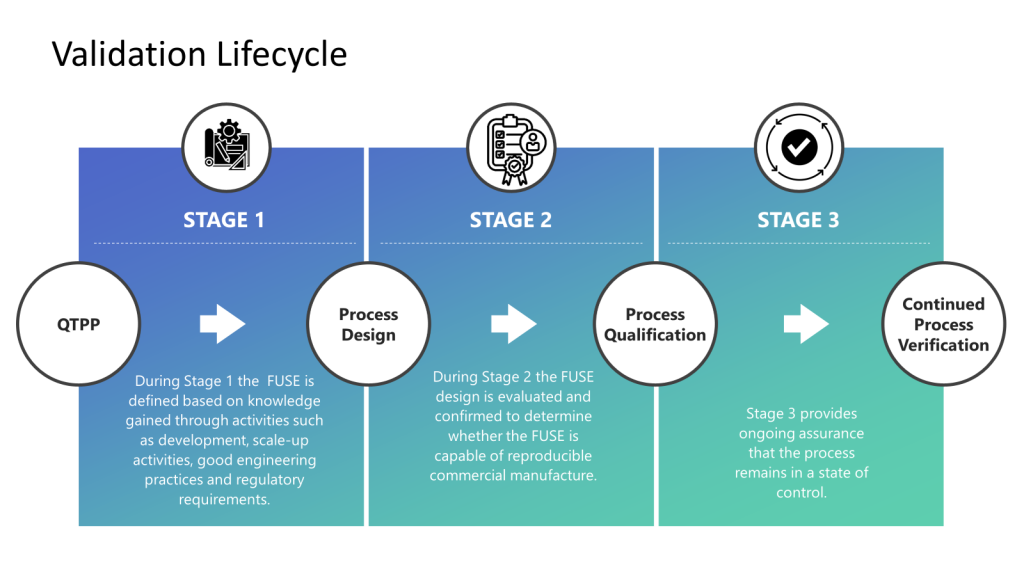
Risk management plays a pivotal role in validation by enabling a risk-based approach to defining validation strategies, ensuring regulatory compliance, mitigating product quality and safety risks, facilitating continuous improvement, and promoting cross-functional collaboration. Integrating risk management principles into the validation lifecycle is essential for maintaining control and consistently producing high-quality products in regulated industries such as biotech and medical devices.

We will conduct various risk assessments in our process lifecycle—many ad hoc (static) and a few living (dynamic). Understanding how they fit together in a larger activity set is crucial.
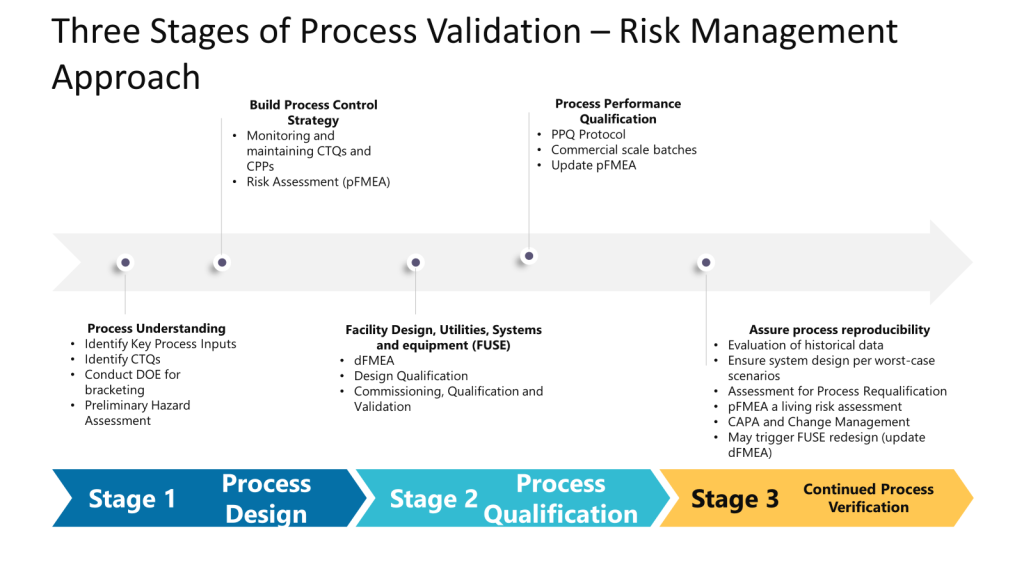
In the Facility, Utilities, Systems, and Equipment (FUSE) space, we are taking the process understanding, translating it into a design, and then performing Design Qualification (DQ) to verify that the critical aspects (CAs) and critical design elements (CDEs) necessary to control risks identified during the quality risk assessment (QRA) are present in the design. This helps mitigate risks to product quality and patient safety. To do this, we need to properly understand the process. Unfortunately, we often start with design before understanding the process and then need to go back and perform rework. Too often I see a dFMEA ignored or as an input to the pFMEA instead of working together in a full risk management cycle.
The Preliminary Hazard Analysis (PHA) supports a pFMEA, which supports a dFMEA, which supports the pFMEA (which also benefits at this stage from a HAACP). Tools fit together to provide the approach. Tools do not become the approach.
Design and Process FMEAs
DFMEA (Design Failure Mode and Effects Analysis) and PFMEA (Process Failure Mode and Effects Analysis) are both methodologies used within the broader FMEA framework to identify and mitigate potential failures. Still, they focus on different aspects of development and manufacturing.
| DFMEA | PFMEA | |
| Scope and Focus | Primarily scrutinizes design to preempt flaws. | Focuses on processes to ensure effectiveness, efficiency and reliability. |
| Stakeholder Involvement | Engages design-oriented teams like engineering, quality engineers, and reliability engineers. | Involves operation-centric personnel such as manufacturing, quality control, quality operations, and process engineers. |
| Inputs and Outputs | Relies on design requirements, product specs, and component interactions to craft a robust product. | Utilizes process steps, equipment capabilities, and parameters to design a stable operational process. |
| Stages in lifecycle | Conducted early in development, concurrent with the design phase, it aids in early issue detection and minimizes design impact. | Executed in production planning post-finalized design, ensuring optimized operations prior to full-scale production. |
| Updated When | Executed in production planning post-finalized design, ensuring optimized operations before full-scale production. | Process changes and under annual review. |
Risk Analysis in the Design Phase
The design qualification phase is especially suitable for determining risks for products and patients stemming from the equipment or machine. These risks should be identified during the design qualification and reflected by appropriate measures in the draft design so that the operator can effectively eliminate, adequately control, and monitor or observe them. To identify design defects (mechanical) or in the creation of systems (electronics) on time and to eliminate them at a low cost, it is advisable to perform the following risk analysis activities for systems, equipment, or processes:
- Categorize the GMP criticality and identify the critical quality attributes and process parameters;
- Categorize the requirements regarding the patient impact and product impact (for example, in the form of a trace matrix);
- Identify critical functions and system elements (e.g., the definition of a calibration concept and preventive maintenance);
- Investigate functions for defect recognition. This includes checking alarms and fault indications, operator error, etc. The result of this risk analysis may be the definition of further maintenance activities, a different assessment of a measurement point, or the identification of topics to include in the operating manuals or procedures.
Additional risk analyses for verifying the design may include usability studies using equipment mock-ups or preliminary production trials (engineering studies) regarding selected topics to prove the feasibility of specific design aspects (e.g., interaction between machine and materials).
Too often, we misunderstand risk assessments and start doing them at the most granular level. This approach allows us to right-size our risk assessments and holistically look at the entire lifecycle.