At the heart of achieving consistent pharmaceutical quality lies operational stability—a fundamental concept that forms the critical middle layer in the House of Quality model. Operational stability serves as the bridge between cultural foundations and the higher-level outcomes of effectiveness, efficiency, and excellence. This critical positioning makes it worthy of detailed examination, particularly as regulatory bodies increasingly emphasize Quality Management Maturity (QMM) as a framework for evaluating pharmaceutical operations.
Understanding Operational Stability in Pharmaceutical Manufacturing
Operational stability represents the state where manufacturing and quality processes exhibit consistent, predictable performance over time with minimal unexpected variations. It refers to the capability of production systems to maintain control within defined parameters regardless of routine challenges that may arise. In pharmaceutical manufacturing, operational stability encompasses everything from batch-to-batch consistency to equipment reliability, from procedural adherence to supply chain resilience.
The essence of operational stability lies in its emphasis on reliability and predictability. A stable operation delivers consistent outcomes not by chance but by design—through robust systems that can withstand normal operating stresses without performance degradation. Pharmaceutical operations that achieve stability demonstrate the ability to maintain critical quality attributes within specified limits while accommodating normal variability in inputs such as raw materials, human operations, and environmental conditions.
According to the House of Quality model for pharmaceutical quality frameworks, operational stability occupies a central position between cultural foundations and higher-level performance outcomes. This positioning is not accidental—it recognizes that stability is both dependent on cultural excellence below it and necessary for the efficiency and effectiveness that lead to excellence above it.
The Path to Obtaining Operational Stability
Achieving operational stability requires a systematic approach that addresses several interconnected dimensions. This pursuit begins with establishing robust processes designed with sufficient control mechanisms and clear operating parameters. Process design should incorporate quality by design principles, ensuring that processes are inherently capable of consistent performance rather than relying on inspection to catch deviations.
Standard operating procedures form the backbone of operational stability. These procedures must be not merely documented but actively maintained, followed, and continuously improved. This principle applies broadly—authoritative documentation precedes execution, ensuring alignment and clarity.
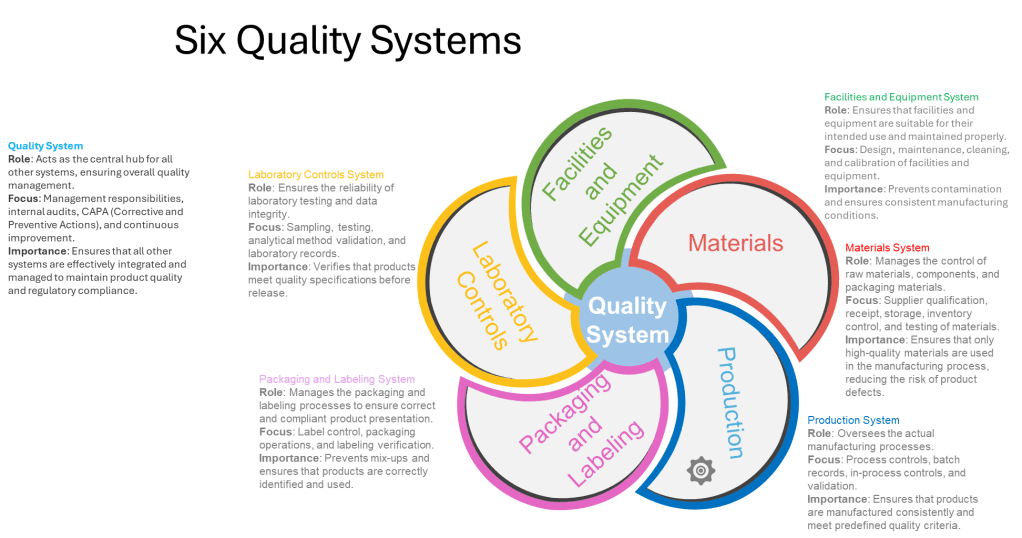
Equipment reliability programs represent another critical component in achieving operational stability. Preventive maintenance schedules, calibration programs, and equipment qualification processes all contribute to ensuring that physical assets support rather than undermine stability goals. The FDA’s guidance on pharmaceutical CGMP regulation emphasizes the importance of the Facilities and Equipment System, which ensures that facilities and equipment are suitable for their intended use and maintained properly.
Supplier qualification and management play an equally important role. As pharmaceutical manufacturing becomes increasingly globalized, with supply chains spanning multiple countries and organizations, the stability of supplied materials becomes essential for operational stability. “Supplier Reliability” appears in the House of Quality model at the same level as operational stability, underscoring their interconnected nature1. Robust supplier qualification programs, ongoing monitoring, and collaborative relationships with key vendors all contribute to supply chain stability that supports overall operational stability.
Human factors cannot be overlooked in the pursuit of operational stability. Training programs, knowledge management systems, and appropriate staffing levels all contribute to consistent human performance. The establishment of a “zero-defect culture” underscores the importance of human factors in achieving true operational stability.
Measuring Operational Stability: Key Metrics and Approaches
Measurement forms the foundation of any improvement effort. For operational stability, measurement approaches must capture both the state of stability and the factors that contribute to it. The pharmaceutical industry utilizes several key metrics to assess operational stability, ranging from process-specific measurements to broader organizational indicators.
Process capability indices (Cp, Cpk) provide quantitative measures of a process’s ability to meet specifications consistently. These statistical measures compare the natural variation in a process against specified tolerances. A process with high capability indices demonstrates the stability necessary for consistent output. These measures help distinguish between common cause variations (inherent to the process) and special cause variations (indicating potential instability).
Deviation rates and severity classification offer another window into operational stability. Tracking not just the volume but the nature and significance of deviations provides insight into systemic stability issues. The following table outlines how different deviation patterns might be interpreted:
| Deviation Pattern | Stability Implication | Recommended Response |
|---|---|---|
| Low frequency, low severity | Good operational stability | Continue monitoring, seek incremental improvements |
| Low frequency, high severity | Critical vulnerability despite apparent stability | Root cause analysis, systemic preventive actions |
| High frequency, low severity | Degrading stability, risk of normalization of deviance | Process review, operator training, standard work reinforcement |
| High frequency, high severity | Fundamental stability issues | Comprehensive process redesign, management system review |
Equipment reliability metrics such as Mean Time Between Failures (MTBF) and Overall Equipment Effectiveness (OEE) provide visibility into the physical infrastructure supporting operations. These measures help identify whether equipment-related issues are undermining otherwise well-designed processes.
Batch cycle time consistency represents another valuable metric for operational stability. In stable operations, the time required to complete batch manufacturing should fall within a predictable range. Increasing variability in cycle times often serves as an early warning sign of degrading operational stability.
Right-First-Time (RFT) batch rates measure the percentage of batches that proceed through the entire manufacturing process without requiring rework, deviation management, or investigation. High and consistent RFT rates indicate strong operational stability.
Leveraging Operational Stability for Organizational Excellence
Once achieved, operational stability becomes a powerful platform for broader organizational excellence. Robust operational stability delivers substantial business benefits that extend throughout the organization.
Resource optimization represents one of the most immediate benefits. Stable operations require fewer resources dedicated to firefighting, deviation management, and rework. This allows for more strategic allocation of both human and financial resources. As noted in the St. Gallen reports “organizations with higher levels of cultural excellence, including employee engagement and continuous improvement mindsets supports both quality and efficiency improvements.”
Stable operations enable focused improvement efforts. Rather than dispersing improvement resources across multiple priority issues, organizations can target specific opportunities for enhancement. This focused approach yields more substantial gains and allows for the systematic building of capabilities over time.
Regulatory confidence grows naturally from demonstrated operational stability. Regulatory agencies increasingly look beyond mere compliance to assess the maturity of quality systems. The FDA’s Quality Management Maturity (QMM) program explicitly recognizes that mature quality systems are characterized by consistent, reliable processes that ensure quality objectives and promote continual improvement.
Market differentiation emerges as companies leverage their operational stability to deliver consistently high-quality products with reliable supply. In markets where drug shortages have become commonplace, the ability to maintain stable supply becomes a significant competitive advantage.
Innovation capacity expands when operational stability frees resources and attention previously consumed by basic operational problems. Organizations with stable operations can redirect energy toward innovation in products, processes, and business models.
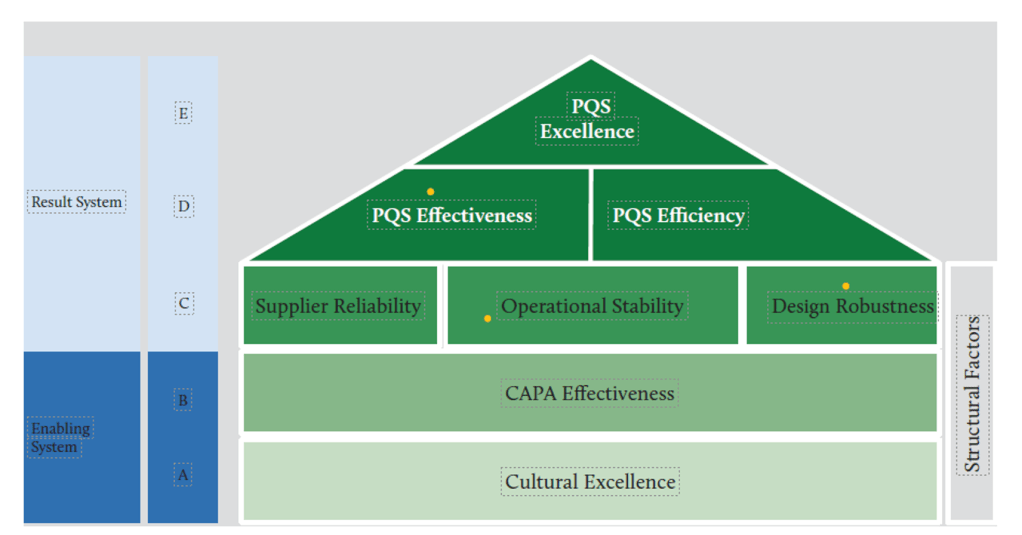
Operational Stability within the House of Quality Model
The House of Quality model places operational stability in a pivotal middle position. This architectural metaphor is instructive—like the middle floors of a building, operational stability both depends on what lies beneath it and supports what rises above it. Understanding this positioning helps clarify operational stability’s role in the broader quality management system.
Cultural excellence lies at the foundation of the House of Quality. This foundation provides the mindset, values, and behaviors necessary for sustained operational stability. Without this cultural foundation, attempts to establish operational stability will likely prove short-lived. At a high level of quality management maturity, organizations operate optimally with clear signals of alignment, where quality and risk management stem from and support the organization’s objectives and values.
Above operational stability in the House of Quality model sit Effectiveness and Efficiency, which together lead to Excellence at the apex. This positioning illustrates that operational stability serves as the essential platform enabling both effectiveness (doing the right things) and efficiency (doing things right). Research from the St. Gallen reports found that “plants with more effective quality systems also tend to be more efficient in their operations,” although “effectiveness only explained about 4% of the variation in efficiency scores.”
The House of Quality model also places Supplier Reliability and Design Robustness at the same level as Operational Stability. This horizontal alignment stems from these three elements work in concert as the critical middle layer of the quality system. Collectively, they provide the stable platform necessary for higher-level performance.
| Element | Relationship to Operational Stability | Joint Contribution to Upper Levels |
|---|---|---|
| Supplier Reliability | Provides consistent input materials essential for operational stability | Enables predictable performance and resource optimization |
| Operational Stability | Creates consistent process performance regardless of normal variations | Establishes the foundation for systematic improvement and performance optimization |
| Design Robustness | Ensures processes and products can withstand variation without quality impact | Reduces the resource burden of controlling variation, freeing capacity for improvement |
The Critical Middle: Why Operational Stability Enables PQS Effectiveness and Efficiency
Operational stability functions as the essential bridge between cultural foundations and higher-level performance outcomes. This positioning highlights its critical role in translating quality culture into tangible quality performance.
Operational stability enables PQS effectiveness by creating the conditions necessary for systems to function as designed. The PQS effectiveness visible in the upper portions of the House of Quality depends on reliable execution of core processes. When operations are unstable, even well-designed quality systems fail to deliver their intended outcomes.
Operational stability enables efficiency by reducing wasteful activities associated with unstable processes. Without stability, efficiency initiatives often fail to deliver sustainable results as resources continue to be diverted to managing instability.
The relationship between operational stability and the higher levels of the House of Quality follows a hierarchical pattern. Attempts to achieve efficiency without first establishing stability typically result in fragile systems that deliver short-term gains at the expense of long-term performance. Similarly, effectiveness cannot be sustained without the foundation of stability. The model implies a necessary sequence: first cultural excellence, then operational stability (alongside supplier reliability and design robustness), followed by effectiveness and efficiency, ultimately leading to excellence.
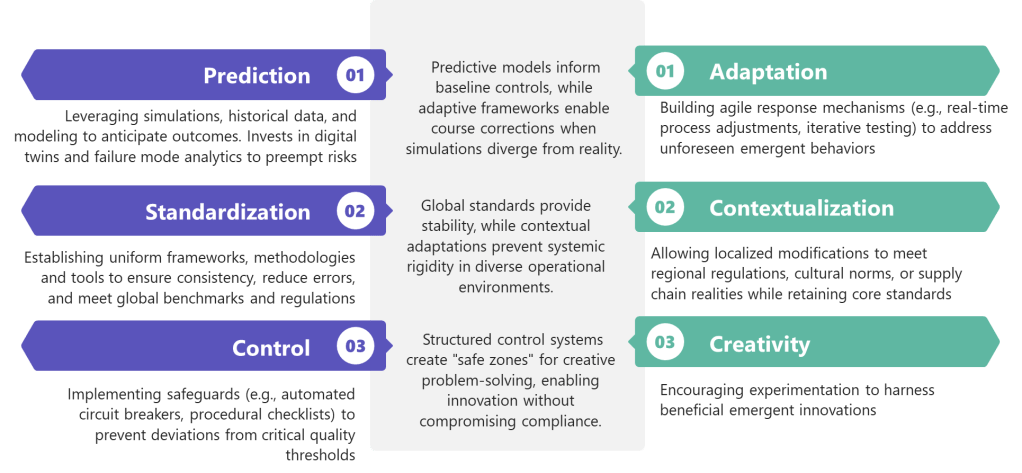
Balancing Operational Stability with Innovation and Adaptability
While operational stability provides numerous benefits, it must be balanced with innovation and adaptability to avoid organizational rigidity. There is a potential negative consequences of an excessive focus on efficiency, including reduced resilience and flexibility which can lead to stifled innovation and creativity.
The challenge lies in establishing sufficient stability to enable consistent performance while maintaining the adaptability necessary for continuous improvement and innovation. This balance requires thoughtful design of stability mechanisms, ensuring they control critical quality attributes without unnecessarily constraining beneficial innovation.
Process characterization plays an important role in striking this balance. By thoroughly understanding which process parameters truly impact critical quality attributes, organizations can focus stability efforts where they matter most while allowing flexibility elsewhere. This selective approach to stability creates what might be called “bounded flexibility”—freedom to innovate within well-understood boundaries.
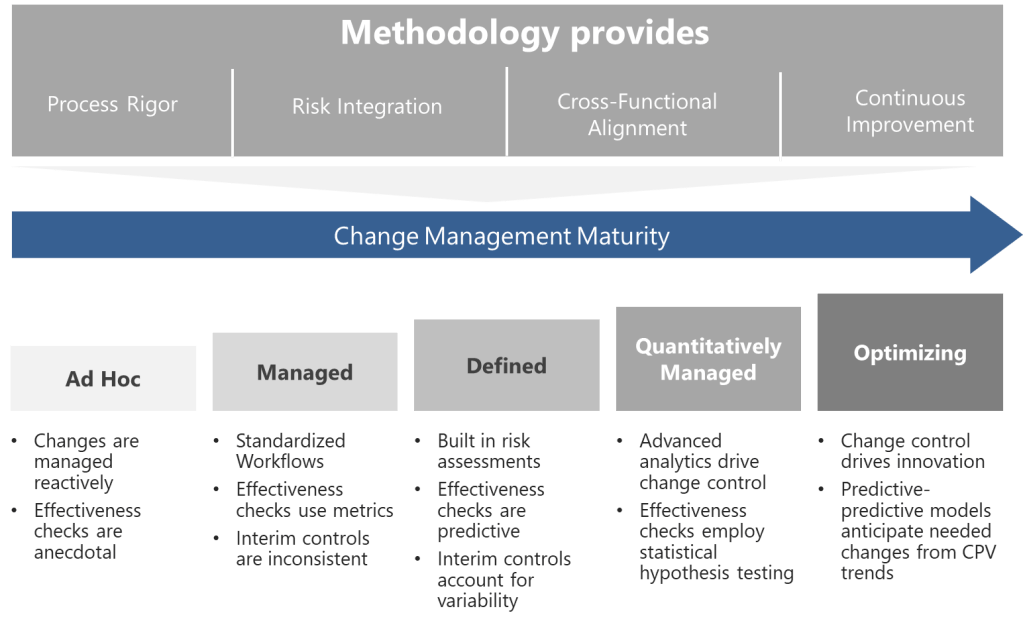
Change management systems represent another critical mechanism for balancing stability with innovation. Well-designed change management ensures that innovations are implemented in a controlled manner that preserves operational stability. ICH Q10 specifically identifies Change Management Systems as a key element of the Pharmaceutical Quality System, emphasizing its importance in maintaining this balance.
Measuring Quality Management Maturity through Operational Stability
Regulatory agencies increasingly recognize operational stability as a key indicator of Quality Management Maturity (QMM). The FDA’s QMM program evaluates organizations across multiple dimensions, with operational performance being a central consideration.
Organizations can assess their own QMM level by examining the nature and pattern of their operational stability. The following table presents a maturity progression framework related to operational stability:
| Maturity Level | Operational Stability Characteristics | Evidence Indicators |
|---|---|---|
| Reactive (Level 1) | Unstable processes requiring constant intervention | High deviation rates, frequent batch rejections, unpredictable cycle times |
| Controlled (Level 2) | Basic stability achieved through rigid controls and extensive oversight | Low deviation rates but high oversight costs, limited process understanding |
| Predictive (Level 3) | Processes demonstrate inherent stability with normal variation understood | Statistical process control effective, leading indicators utilized |
| Proactive (Level 4) | Stability maintained through systemic approaches rather than individual efforts | Root causes addressed systematically, culture of ownership evident |
| Innovative (Level 5) | Stability serves as platform for continuous improvement and innovation | Stability metrics consistently excellent, resources focused on value-adding activities |
This maturity progression aligns with the FDA’s emphasis on QMM as “the state attained when drug manufacturers have consistent, reliable, and robust business processes to achieve quality objectives and promote continual improvement”.
Practical Approaches to Building Operational Stability
Building operational stability requires a comprehensive approach addressing process design, organizational capabilities, and management systems. Several practical methods have proven particularly effective in pharmaceutical manufacturing environments.
Statistical Process Control (SPC) provides a systematic approach to monitoring processes and distinguishing between common cause and special cause variation. By establishing control limits based on natural process variation, SPC helps identify when processes are operating stably within expected variation versus when they experience unusual variation requiring investigation. This distinction prevents over-reaction to normal variation while ensuring appropriate response to significant deviations.
Process validation activities establish scientific evidence that a process can consistently deliver quality products. Modern validation approaches emphasize ongoing process verification rather than point-in-time demonstrations, aligning with the continuous nature of operational stability.
Root cause analysis capabilities ensure that when deviations occur, they are investigated thoroughly enough to identify and address underlying causes rather than symptoms. This prevents recurrence and systematically improves stability over time. The CAPA (Corrective Action and Preventive Action) system plays a central role in this aspect of building operational stability.
Knowledge management systems capture and make accessible the operational knowledge that supports stability. By preserving institutional knowledge and making it available when needed, these systems reduce dependence on individual expertise and create more resilient operations. This aligns with ICH Q10’s emphasis on “expanding the body of knowledge”.
Training and capability development ensure that personnel possess the necessary skills to maintain operational stability. Investment in operator capabilities pays dividends through reduced variability in human performance, often a significant factor in overall operational stability.
Operational Stability as the Engine of Quality Excellence
Operational stability occupies a pivotal position in the House of Quality model—neither the foundation nor the capstone, but the essential middle that translates cultural excellence into tangible performance outcomes. Its position reflects its dual nature: dependent on cultural foundations for sustainability while enabling the effectiveness and efficiency that lead to excellence.
The journey toward operational stability is not merely technical but cultural and organizational. It requires systematic approaches, appropriate metrics, and balanced objectives that recognize stability as a means rather than an end. Organizations that achieve robust operational stability position themselves for both regulatory confidence and market leadership.
As regulatory frameworks evolve toward Quality Management Maturity models, operational stability will increasingly serve as a differentiator between organizations. Those that establish and maintain strong operational stability will find themselves well-positioned for both compliance and competition in an increasingly demanding pharmaceutical landscape.
The House of Quality model provides a valuable framework for understanding operational stability’s role and relationships. By recognizing its position between cultural foundations and performance outcomes, organizations can develop more effective strategies for building and leveraging operational stability. The result is a more robust quality system capable of delivering not just compliance but true quality excellence that benefits patients, practitioners, and the business itself.