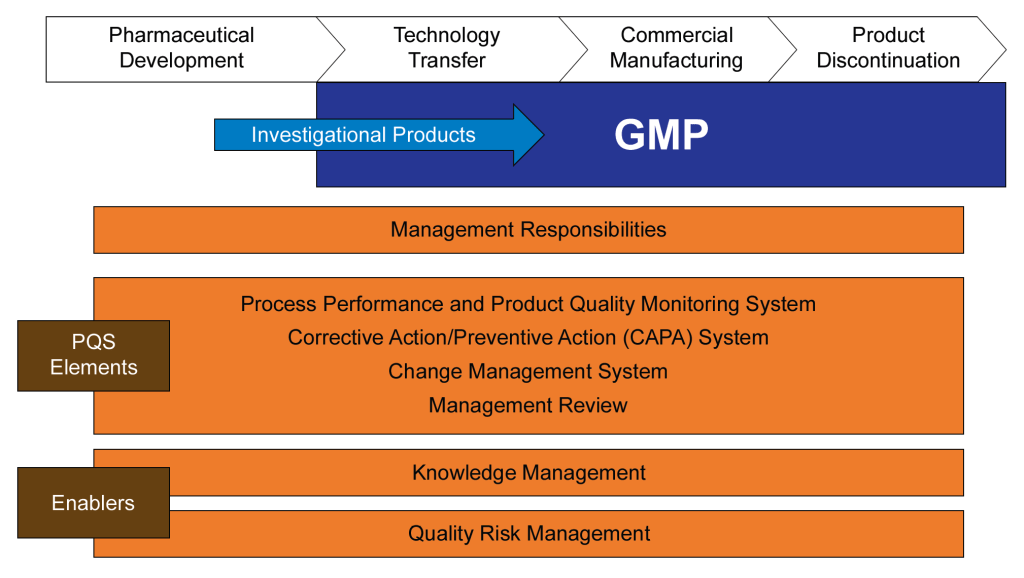
Layers of Controls Analysis (LOCA)
Layers of Controls Analysis (LOCA) provides a comprehensive framework for evaluating multiple layers of protection to reduce and manage operational risks. By examining both preventive and mitigative control measures simultaneously, LOCA allows organizations to gain a holistic view of their risk management strategy. This approach is particularly valuable in complex operational environments where multiple safeguards and protective systems are in place.
One of the key strengths of LOCA is its ability to identify gaps in protection. By systematically analyzing each layer of control, from basic process design to emergency response procedures, LOCA can reveal areas where additional safeguards may be necessary. This insight is crucial for guiding decisions on implementing new risk reduction measures or enhancing existing ones. The analysis helps organizations prioritize their risk management efforts and allocate resources more effectively.
Furthermore, LOCA provides a structured way to document and justify risk reduction measures. This documentation is invaluable for regulatory compliance, internal audits, and continuous improvement initiatives. By clearly outlining the rationale behind each protective layer and its contribution to overall risk reduction, organizations can demonstrate due diligence in their safety and risk management practices.
Another significant advantage of LOCA is its promotion of a holistic view of risk control. Rather than evaluating individual safeguards in isolation, LOCA considers the cumulative effect of multiple protective layers. This approach recognizes that risk reduction is often achieved through the interaction of various control measures, ranging from engineered systems to administrative procedures and emergency response capabilities.
By building on other risk assessment techniques, such as Hazard and Operability (HAZOP) studies and Fault Tree Analysis, LOCA provides a more complete picture of protection systems. It allows organizations to assess the effectiveness of their entire risk management strategy, from prevention to mitigation, and ensures that risks are reduced to an acceptable level. This comprehensive approach is particularly valuable in high-hazard industries where the consequences of failures can be severe.
LOCA combines elements of two other methods – Layers of Protection Analysis (LOPA) and Layers of Mitigation Analysis (LOMA).
Layers of Protection Analysis
To execute a Layers of Protection Analysis (LOPA), follow these key steps:
Define the hazardous scenario and consequences:
- Clearly identify the hazardous event being analyzed
- Determine the potential consequences if all protection layers fail
Identify initiating events:
- List events that could trigger the hazardous scenario
- Estimate the frequency of each initiating event
Identify Independent Protection Layers (IPLs):
- Determine existing safeguards that can prevent the scenario
- Evaluate if each safeguard qualifies as an IPL (independent, auditable, effective)
- Estimate the Probability of Failure on Demand (PFD) for each IPL
Identify Conditional Modifiers:
- Determine factors that impact scenario probability (e.g. occupancy, ignition probability)
- Estimate probability for each modifier
Calculate scenario frequency:
- Multiply initiating event frequency by PFDs of IPLs and conditional modifiers
Compare to risk tolerance criteria:
- Determine if calculated frequency meets acceptable risk level
- If not, identify need for additional IPLs
Document results:
- Record all assumptions, data sources, and calculations
- Summarize findings and recommendations
Review and validate:
- Have results reviewed by subject matter experts
- Validate key assumptions and data inputs
Key aspects for successful LOPA execution
- Use a multidisciplinary team
- Ensure independence between IPLs
- Be conservative in estimates
- Focus on prevention rather than mitigation
- Consider human factors in IPL reliability
- Use consistent data sources and methods
Layers of Mitigation Analysis
LOMA focuses on analyzing reactionary or mitigative measures, as opposed to preventive measures.
A LOCA as part of Contamination Control
A Layers of Controls Analysis (LOCA) can be effectively applied to contamination control in biotech manufacturing by systematically evaluating multiple layers of protection against contamination risks.

To determine potential hazards when conducting a Layer of Controls Analysis (LOCA) for contamination control in biotech, follow these steps:
- Form a multidisciplinary team: Include members from manufacturing, quality control, microbiology, engineering, and environmental health & safety to gain diverse perspectives.
- Review existing processes and procedures: Examine standard operating procedures, experimental protocols, and equipment manuals to identify potential risks associated with each step.
- Consider different hazard types. Focus on categories like:
- Biological hazards (e.g., microorganisms, cell lines)
- Chemical hazards (e.g., toxic substances, flammable materials)
- Physical hazards (e.g., equipment-related risks)
- Radiological hazards (if applicable)
- Analyze specific contamination hazard types for biotech settings:
- Mix-up: Materials used for the wrong product
- Mechanical transfer: Cross-contamination via personnel, supplies, or equipment
- Airborne transfer: Contaminant movement through air/HVAC systems
- Retention: Inadequate removal of materials from surfaces
- Proliferation: Potential growth of biological agents
- Conduct a process analysis: Break down each laboratory activity into steps and identify potential hazards at each stage.
- Consider human factors: Evaluate potential for human error, such as incorrect handling of materials or improper use of equipment.
- Assess facility and equipment: Examine the layout, containment measures, and equipment condition for potential hazards.
- Review past incidents and near-misses: Analyze previous safety incidents or close calls to identify recurring or potential hazards.
- Consult relevant guidelines and regulations: Reference industry standards, biosafety guidelines, and regulatory requirements to ensure comprehensive hazard identification.
- Use brainstorming techniques: Encourage team members to think creatively about potential hazards that may not be immediately obvious.
- Evaluate hazards at different scales: Consider how hazards might change as processes scale up from research to production levels.

- Facility Design and Engineering Controls
- Cleanroom design and classification
- HVAC systems with HEPA filtration
- Airlocks and pressure cascades
- Segregated manufacturing areas
- Equipment and Process Design
- Closed processing systems
- Single-use technologies
- Sterilization and sanitization systems
- In-line filtration
- Operational Controls
- Aseptic techniques and procedures
- Environmental monitoring programs
- Cleaning and disinfection protocols
- Personnel gowning and hygiene practices
- Quality Control Measures
- In-process testing (e.g., bioburden, endotoxin)
- Final product sterility testing
- Environmental monitoring data review
- Batch record review
- Organizational Controls
- Training programs
- Standard operating procedures (SOPs)
- Quality management systems
- Change control processes
- Evaluate reliability and capability of each control:
- Review historical performance data for each control measure
- Assess the control’s ability to prevent or detect contamination
- Consider the control’s consistency in different operating conditions
- Consider potential failure modes:
- Conduct a Failure Mode and Effects Analysis (FMEA) for each control
- Identify potential ways the control could fail or be compromised
- Assess the likelihood and impact of each failure mode
- Evaluate human factors:
- Assess the complexity and potential for human error in each control
- Review training effectiveness and compliance with procedures
- Consider ergonomics and usability of equipment and systems
- Analyze technology effectiveness:
- Evaluate the performance of automated systems and equipment
- Assess the reliability of monitoring and detection technologies
- Consider the integration of different technological controls
- Quantify risk reduction:
- Assign risk reduction factors to each layer based on its effectiveness
- Use a consistent scale (e.g., 1-10) to rate each control’s risk reduction capability
- Calculate the cumulative risk reduction across all layers
- Assess interdependencies between layers:
- Identify any controls that rely on or affect other controls
- Evaluate how failures in one layer might impact the effectiveness of others
- Consider potential common mode failures across multiple layers
- Review control performance metrics:
- Analyze trends in environmental monitoring data
- Examine out-of-specification results and their root causes
- Assess the frequency and severity of contamination events
- Determine acceptable risk levels:
- Define your organization’s risk tolerance for contamination events
- Compare current risk levels against these thresholds
- Identify gaps:
- Highlight areas where current controls fall short of required protection
- Note processes or areas with insufficient redundancy
- Propose improvements:
- Suggest enhancements to existing controls
- Recommend new control measures to address identified gaps
- Prioritize actions:
- Rank proposed improvements based on risk reduction potential and feasibility
- Consider cost-benefit analysis for major changes
- Seek expert input:
- Consult with subject matter experts on proposed improvements
- Consider third-party assessments for critical areas
- Plan for implementation:
- Develop action plans for addressing identified gaps
- Assign responsibilities and timelines for improvements

- Document and review:
- Create a comprehensive LOCA document
- Regularly review and update the analysis. This is a living risk assessment.
- Incorporate lessons learned from any contamination events

- Implement continuous monitoring and review:
- Establish key performance indicators (KPIs)
- Conduct regular audits and inspections
- Review environmental monitoring data trends
- Develop a holistic CCS document:
- Describe overall contamination control approach
- Detail how different controls work together
- Include risk assessments and rationales
- Establish governance and oversight:
- Create a cross-functional CCS team
- Define roles and responsibilities
- Implement a regular review process
- Integrate with quality systems:
- Align CCS with existing quality management processes
- Ensure change control procedures consider CCS impact
- Provide comprehensive training:
- Train all personnel on CCS principles and practices
- Implement contamination control ambassador program

- Implement regular review cycles:
- Schedule periodic reviews of the LOCA (e.g., annually or bi-annually)
- Involve a cross-functional team including quality, manufacturing, and engineering
- Analyze trends and data:
- Review environmental monitoring data
- Examine out-of-specification results and their root causes
- Assess the frequency and severity of contamination events
- Identify improvement opportunities:
- Use gap analysis to compare current controls against industry best practices
- Evaluate new technologies and methodologies for contamination control
- Consider feedback from contamination control ambassadors and staff
- Prioritize improvements:
- Rank proposed enhancements based on risk reduction potential and feasibility
- Consider cost-benefit analysis for major changes
- Implement changes:
- Update standard operating procedures (SOPs) as needed
- Provide training on new or modified control measures
- Validate changes to ensure effectiveness
- Monitor and measure impact:
- Establish key performance indicators (KPIs) for each layer of control
- Track improvements in contamination rates and overall control effectiveness
- Foster a culture of continuous improvement:
- Encourage proactive reporting of potential issues
- Recognize and reward staff contributions to contamination control
- Stay updated on regulatory requirements:
- Regularly review and incorporate changes in regulations (e.g., EU GMP Annex 1)
- Attend industry conferences and workshops on contamination control
- Integrate with overall quality systems:
- Ensure LOCA improvements align with the site’s Quality Management System
- Update the Contamination Control Strategy (CCS) document as needed
- Leverage technology:
- Implement digital solutions for environmental monitoring and data analysis
- Consider advanced technologies like rapid microbial detection methods
- Conduct periodic audits:
- Perform surprise audits to ensure adherence to protocols
- Use findings to further refine the LOCA and control measures

{kind=link}
{kind=link}