Seriously, not much surprising here. What this guide definitely does is place early research in the framework of Q10 and point out that there is one quality system to rule them all and that level of rigor is based on risk.
The yearly database of 483s has been updated by the FDA. Lots will be written on themes as we end the year, so I decided to give a few of my immediate observations.
I find that over time what I focus in on changes, as my jobs evolve and change, and the interests I have shift. However, some things never change, so let us talk change.
Reference Number
Short Description
Long Description
2020 Frequency
2019 Frequency
2018 Frequency
21 CFR 211.100(a)
Changes to Procedures Not Reviewed, Approved
Changes to written procedures are not [drafted, reviewed and approved by the appropriate organizational unit] [reviewed and approved by the quality control unit]. Specifically, ***
8
13
9
21 CFR 211.160(a)
Lab controls established, including changes
The establishment of [specifications] [standards] [sampling plans] [test procedures] [laboratory control mechanisms] including any changes thereto, are not [drafted by the appropriate organizational unit] [reviewed and approved by the quality control unit]. Specifically, ***
4
18
17
21 CFR 212.20(c)
Adverse effects of changes made
You did not demonstrate that any change does not adversely affect the [identity] [strength] [quality] [purity] of your PET drug. Specifically,***
1
1
1
483s related to changes
I think its fair to say the decreases as a result of the pandemic and the reduced inspections.
Over on the device side of things we see:
Reference Number
Short Description
Long Description
Frequency
21 CFR 820.30(i)
Design changes – Lack of or Inadequate Procedures
Procedures for design change have not been [adequately] established. Specifically,***
26
21 CFR 820.40(b)
Document change records, maintained.
Records of changes to documents were not [adequately] maintained. Specifically, ***
6
21 CFR 820.70(b)
Production and Process Change Procedures, lack of or Inad.
Procedures for changes to a [specification] [method] [process] [procedure] have not been [adequately] established. Specifically, ***
5
21 CFR 820.75(c)
Process changes – review, evaluation and revalidation
A validated process was not [reviewed and evaluated] [revalidated] when changes or process deviations occurred. Specifically, ***
5
21 CFR 820.40(b)
Change records, content
Records of changes did not include [a description of the change] [identification of the affected documents] [the signature of the approving official(s)] [the approval date] [when the change became effective]. Specifically, ***
3
21 CFR 820.50(b)
Supplier notification of changes
There is no agreement with [suppliers] [contractors] [consultants] to notify you of changes in the product or service. Specifically, ***
3
21 CFR 820.75(c)
Documentation – review in response to changes or deviations
There is no documentation of the [review and evaluation of a process] [revalidation of a process] performed in response to changes or process deviations. Specifically, ***
1
Device 473s around change
I’m a firm believer that pharma should always pay attention to the medical device side for 483s. A lot of us are combination products now (or will be) and there is always good trends to be aware of.
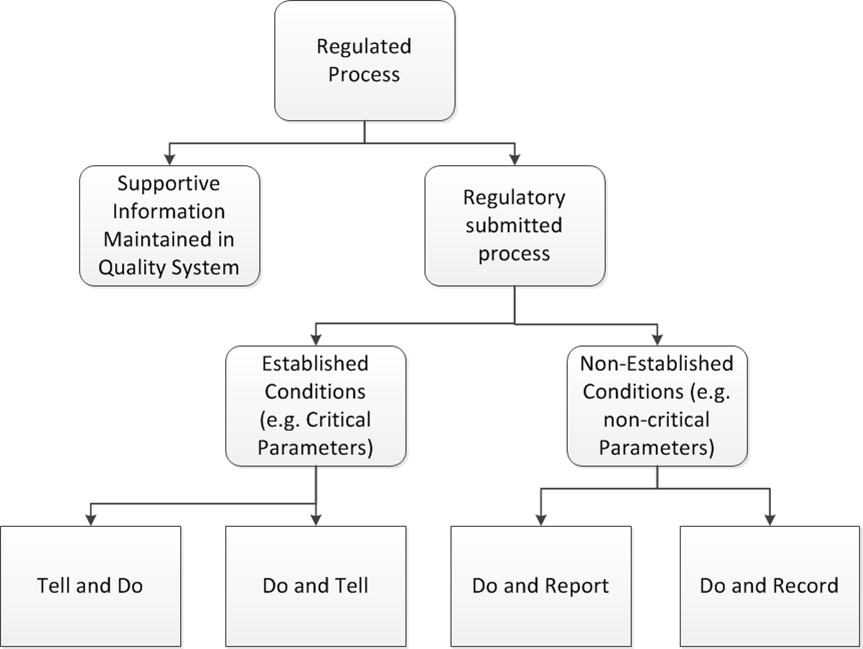
Prior to the adoption of Q12 in Singapore at the end of 2019 there was a lot of rumbling from regulatory agencies on how Q12 would be more aspirational in many ways. In the last few weeks we’ve started to see just what that will mean.
FDA to release a guidance
The FDA’s Mahesh Ramanadham, from the Office of Pharmaceutical Quality in the FDA’s Center for Drug Evaluation and Research, provided an update on the agency’s implementation of ICH Q12 in the US on 25 February at the annual IFPAC meeting in North Bethesda, Md. He started that the FDA will soon be issuing guidance implementing the International Council on Harmonization’s Q12 guideline in the US that will, among other things, translate ICH post-approval change classification categories to FDA supplement categories, and address how to file established conditions (ECs).
This Q12 guidance will replace the agency’s 2015 draft guidance for industry on established conditions and reportable chemistry, manufacturing and controls changes to approved drug and biological products. It is expected to be issued in May 2020. The guidance will also discuss the relationship between FDA comparability protocols and the post-approval change management protocol (PACMP) established by the ICH Q12 guideline.
EU says not so fast in their adoption
However, additional scientific risk-based approaches to defining Established Conditions and associated reporting categories, as described in Chapter 3.2.3, and the Product Lifecycle Management (PLCM) Document, as described in Chapter 5, are not considered compatible with the existing EU legal framework on variations.
It is important to note that the legal framework always takes precedence over technical and scientific guidelines. More specifically this means that the definition of Established Conditions and their reporting categories must follow the requirements laid down in the current EU Variations Regulation and associated EU Variations Guidelines. With respect to the Product Lifecycle Management (PLCM) document, in case such a document is submitted, it cannot be currently recognised in the EU due to the fact that it is not referred to in the EU legal framework.
In an explanatory note accompanying the adoption of ICH Q12 and related annexes, the European Commission and the European Medicines Agency point out that there are “some conceptual differences” between the ICH guideline and the EU legal framework on managing post-approval changes, ie, the variations regulation (Regulation (EC) No 1234/2008).
The EU authorities offer no clarity on when and how ICH Q12 would be fully implemented in the EU. The note merely states that the new “tools and concepts in the ICH Q12 guideline that are not foreseen in the EU legal framework will be considered when this framework will be reviewed.” The EU regulators said they would continue to work on the implementation of the ICH Q12 within the existing EU legal framework. The explanatory note also points out that despite some conceptual differences between ICH Q12 and the EU framework, there is also considerable common ground. In fact, some tools and concepts in ICH Q12 tools can already be applied by industry by following the current EU variations framework.
Next Steps
Companies should be ensuring that their knowledge management and risk management processes and understanding continue to grow. ICH Q12 will be a rocky road and I’m not sure we’ll see some of the potential streamlining of regulatory processes for a long time.
I hear this from colleagues all the time, how do you keep on top of so much news? How have are you always the first person to know this thing?
Keeping on top of trends, of regulatory changes, of new event is critical. This is a big part of content, and from that we bring context. It is part of what makes us Subject Matter Experts.
But let’s be honest, there is a lot out there to stay aware of, and it can eat a lot of unproductive hours up, especially with so much new content on the web being added daily, it can be tough to keep up with what’s happening online. But if you rely on visiting specific websites every day, doing Google searches, or relying on social media to keep them informed you will get overwhelmed and miss important things. The best solution I have found is pretty old-school: The RSS feed.
About every site you need to follow, from a trade publication to a government agency to a professional society to all those blogs publishes an RSS feed (including this one). Often they publish more than one which can compartmentalize the reading and help you narrow down to specific topics that interest you.
You then use a reader to aggregate these feeds into compartments that make sense to you, skim the headlines at your leisure and read those items that seem relevant to you. This gives you a comprehensive, regularly updated look at all the content your favorite sites publish throughout the day. Think of it as the ultimate aggregator; every morsel from every source you care about, fed directly to you.
I use Feedly, but there are several good ones out there, most free to use. A lot of friends tell me I should move to Inoreader.
Do not be afraid of the massive amounts of information out there. Like anything, it’s all about getting the right tool for the job.
Between January 2016 and May 2019, you recorded approximately 397 customer complaints related to container closure issues (e.g., approximately 60%), product separation, lack of effect and adverse events. Your quality unit failed to adequately review these complaints, identify trends, and implement effective CAPAs. During the inspection you explained that these lapses in quality system performance were due to underperforming staff, who had since been dismissed. In your response, you attributed these and other quality related issues to under staffing of the quality unit as your business expanded.
Your response is inadequate because you failed to appropriately address your quality unit not performing their required duties. Your firm must provide the quality unit with the appropriate authority, sufficient resources, and staff to carry out its responsibilities to consistently ensure drug quality.
Continuing the trend of making me petrified about generics, this warning letter is a roller-coaster read. One big set of failures is to actually investigate and apply appropriate resources to the quality unit. And then the management had the gall to blame the employees.
80+ years of quality principles ignored. I personally thought the FDA was being overly nice.
There are basically three questions to answer:
Do you have a properly established, staffed, and managed Quality Unit?
Does your Quality Unit have appropriate responsibilities and authority?
Does your Quality Unit have access to the data it needs to make informed decisions?