Beyond the Shadow of Failure
Problem-solving is too often shaped by the assumption that the system is perfectly understood and fully specified. If something goes wrong—a deviation, a batch out-of-spec, or a contamination event—our approach is to dissect what “failed” and fix that flaw, believing this will restore order. This way of thinking, which I call the malfunction mindset, is as ingrained as it is incomplete. It assumes that successful outcomes are the default, that work always happens as written in SOPs, and that only failure deserves our scrutiny.
But here’s the paradox: most of the time, our highly complex manufacturing environments actually succeed—often under imperfect, shifting, and not fully understood conditions. If we only study what failed, and never question how our systems achieve their many daily successes, we miss the real nature of pharmaceutical quality: it is not the absence of failure, but the presence of robust, adaptive work. Taking this broader, more nuanced perspective is not just an academic exercise—it’s essential for building resilient operations that truly protect patients, products, and our organizations.
Drawing from my thinking through zemblanity (the predictable but often overlooked negative outcomes of well-intentioned quality fixes), the effectiveness paradox (why “nothing bad happened” isn’t proof your quality system works), and the persistent gap between work-as-imagined and work-as-done, this post explores why the malfunction mindset persists, how it distorts investigations, and what future-ready quality management should look like.
The Allure—and Limits—of the Failure Model
Why do we reflexively look for broken parts and single points of failure? It is, as Sidney Dekker has argued, both comforting and defensible. When something goes wrong, you can always point to a failed sensor, a missed checklist, or an operator error. This approach—introducing another level of documentation, another check, another layer of review—offers a sense of closure and regulatory safety. After all, as long as you can demonstrate that you “fixed” something tangible, you’ve fulfilled investigational due diligence.
Yet this fails to account for how quality is actually produced—or lost—in the real world. The malfunction model treats systems like complicated machines: fix the broken gear, oil the creaky hinge, and the machine runs smoothly again. But, as Dekker reminds us in Drift Into Failure, such linear thinking ignores the drift, adaptation, and emergent complexity that characterize real manufacturing environments. The truth is, in complex adaptive systems like pharmaceutical manufacturing, it often takes more than one “error” for failure to manifest. The system absorbs small deviations continuously, adapting and flexing until, sometimes, a boundary is crossed and a problem surfaces.
W. Edwards Deming’s wisdom rings truer than ever: “Most problems result from the system itself, not from individual faults.” A sustainable approach to quality is one that designs for success—and that means understanding the system-wide properties enabling robust performance, not just eliminating isolated malfunctions.
Procedural Fundamentalism: The Work-as-Imagined Trap
One of the least examined, yet most impactful, contributors to the malfunction mindset is procedural fundamentalism—the belief that the written procedure is both a complete specification and an accurate description of work. This feels rigorous and provides compliance comfort, but it is a profound misreading of how work actually happens in pharmaceutical manufacturing.
Work-as-imagined, as elucidated by Erik Hollnagel and others, represents an abstraction: it is how distant architects of SOPs visualize the “correct” execution of a process. Yet, real-world conditions—resource shortages, unexpected interruptions, mismatched raw materials, shifting priorities—force adaptation. Operators, supervisors, and Quality professionals do not simply “follow the recipe”: they interpret, improvise, and—crucially—adjust on the fly.
When we treat procedures as authoritative descriptions of reality, we create the proxy problem: our investigations compare real operations against an imagined baseline that never fully existed. Deviations become automatically framed as problem points, and success is redefined as rigid adherence, regardless of context or outcome.
Complexity, Performance Variability, and Real Success
So, how do pharmaceutical operations succeed so reliably despite the ever-present complexity and variability of daily work?
The answer lies in embracing performance variability as a feature of robust systems, not a flaw. In high-reliability environments—from aviation to medicine to pharmaceutical manufacturing—success is routinely achieved not by demanding strict compliance, but by cultivating adaptive capacity.
Consider environmental monitoring in a sterile suite: The procedure may specify precise times and locations, but a seasoned operator, noticing shifts in people flow or equipment usage, might proactively sample a high-risk area more frequently. This adaptation—not captured in work-as-imagined—actually strengthens data integrity. Yet, traditional metrics would treat this as a procedural deviation.
This is the paradox of the malfunction mindset: in seeking to eliminate all performance variability, we risk undermining precisely those adaptive behaviors that produce reliable quality under uncertainty.
Why the Malfunction Mindset Persists: Cognitive Comfort and Regulatory Reinforcement
Why do organizations continue to privilege the malfunction mindset, even as evidence accumulates of its limits? The answer is both psychological and cultural.
Component breakdown thinking is psychologically satisfying—it offers a clear problem, a specific cause, and a direct fix. For regulatory agencies, it is easy to measure and audit: did the deviation investigation determine the root cause, did the CAPA address it, does the documentation support this narrative? Anything that doesn’t fit this model is hard to defend in audits or inspections.
Yet this approach offers, at best, a partial diagnosis and, at worst, the illusion of control. It encourages organizations to catalog deviations while blindly accepting a much broader universe of unexamined daily adaptations that actually determine system robustness.
Complexity Science and the Art of Organizational Success
To move toward a more accurate—and ultimately more effective—model of quality, pharmaceutical leaders must integrate the insights of complexity science. Drawing from the work of Stuart Kauffman and others at the Santa Fe Institute, we understand that the highest-performing systems operate not at the edge of rigid order, but at the “edge of chaos,” where structure is balanced with adaptability.
In these systems, success and failure both arise from emergent properties—the patterns of interaction between people, procedures, equipment, and environment. The most meaningful interventions, therefore, address how the parts interact, not just how each part functions in isolation.
This explains why traditional root cause analysis, focused on the parts, often fails to produce lasting improvements; it cannot account for outcomes that emerge only from the collective dynamics of the system as a whole.
Investigating for Learning: The Take-the-Best Heuristic
A key innovation needed in pharmaceutical investigations is a shift to what Hollnagel calls Safety-II thinking: focusing on how things go right as well as why they occasionally go wrong.
Here, the take-the-best heuristic becomes crucial. Instead of compiling lists of all deviations, ask: Among all contributing factors, which one, if addressed, would have the most powerful positive impact on future outcomes, while preserving adaptive capacity? This approach ensures investigations generate actionable, meaningful learning, rather than feeding the endless paper chase of “compliance theater.”
Building Systems That Support Adaptive Capability
Taking complexity and adaptive performance seriously requires practical changes to how we design procedures, train, oversee, and measure quality.
- Procedure Design: Make explicit the distinction between objectives and methods. Procedures should articulate clear quality goals, specify necessary constraints, but deliberately enable workers to choose methods within those boundaries when faced with new conditions.
- Training: Move beyond procedural compliance. Develop adaptive expertise in your staff, so they can interpret and adjust sensibly—understanding not just “what” to do, but “why” it matters in the bigger system.
- Oversight and Monitoring: Audit for adaptive capacity. Don’t just track “compliance” but also whether workers have the resources and knowledge to adapt safely and intelligently. Positive performance variability (smart adaptations) should be recognized and studied.
- Quality System Design: Build systematic learning from both success and failure. Examine ordinary operations to discern how adaptive mechanisms work, and protect these capabilities rather than squashing them in the name of “control.”
Leadership and Systems Thinking
Realizing this vision depends on a transformation in leadership mindset—from one seeking control to one enabling adaptive capacity. Deming’s profound knowledge and the principles of complexity leadership remind us that what matters is not enforcing ever-stricter compliance, but cultivating an organizational context where smart adaptation and genuine learning become standard.
Leadership must:
- Distinguish between complicated and complex: Apply detailed procedures to the former (e.g., calibration), but support flexible, principles-based management for the latter.
- Tolerate appropriate uncertainty: Not every problem has a clear, single answer. Creating psychological safety is essential for learning and adaptation during ambiguity.
- Develop learning organizations: Invest in deep understanding of operations, foster regular study of work-as-done, and celebrate insights from both expected and unexpected sources.
Practical Strategies for Implementation
Turning these insights into institutional practice involves a systematic, research-inspired approach:
- Start procedure development with observation of real work before specifying methods. Small scale and mock exercises are critical.
- Employ cognitive apprenticeship models in training, so that experience, reasoning under uncertainty, and systems thinking become core competencies.
- Begin investigations with appreciative inquiry—map out how the system usually works, not just how it trips up.
- Measure leading indicators (capacity, information flow, adaptability) not just lagging ones (failures, deviations).
- Create closed feedback loops for corrective actions—insisting every intervention be evaluated for impact on both compliance and adaptive capacity.
Scientific Quality Management and Adaptive Systems: No Contradiction
The tension between rigorous scientific quality management (QbD, process validation, risk management frameworks) and support for adaptation is a false dilemma. Indeed, genuine scientific quality management starts with humility: the recognition that our understanding of complex systems is always partial, our controls imperfect, and our frameworks provisional.
A falsifiable quality framework embeds learning and adaptation at its core—treating deviations as opportunities to test and refine models, rather than simply checkboxes to complete.
The best organizations are not those that experience the fewest deviations, but those that learn fastest from both expected and unexpected events, and apply this knowledge to strengthen both system structure and adaptive capacity.
Embracing Normal Work: Closing the Gap
Normal pharmaceutical manufacturing is not the story of perfect procedural compliance; it’s the story of people, working together to achieve quality goals under diverse, unpredictable, and evolving conditions. This is both more challenging—and more rewarding—than any plan prescribed solely by SOPs.
To truly move the needle on pharmaceutical quality, organizations must:
- Embrace performance variability as evidence of adaptive capacity, not just risk.
- Investigate for learning, not blame; study success, not just failure.
- Design systems to support both structure and flexible adaptation—never sacrificing one entirely for the other.
- Cultivate leadership that values humility, systems thinking, and experimental learning, creating a culture comfortable with complexity.
This approach will not be easy. It means questioning decades of compliance custom, organizational habit, and intellectual ease. But the payoff is immense: more resilient operations, fewer catastrophic surprises, and, above all, improved safety and efficacy for the patients who depend on our products.
The challenge—and the opportunity—facing pharmaceutical quality management is to evolve beyond compliance theater and malfunction thinking into a new era of resilience and organizational learning. Success lies not in the illusory comfort of perfectly executed procedures, but in the everyday adaptations, intelligent improvisation, and system-level capabilities that make those successes possible.
The call to action is clear: Investigate not just to explain what failed, but to understand how, and why, things so often go right. Protect, nurture, and enhance the adaptive capacities of your organization. In doing so, pharmaceutical quality can finally become more than an after-the-fact audit; it will become the creative, resilient capability that patients, regulators, and organizations genuinely want to hire.
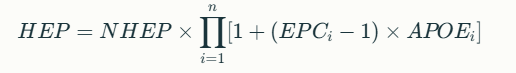






{kind=link}