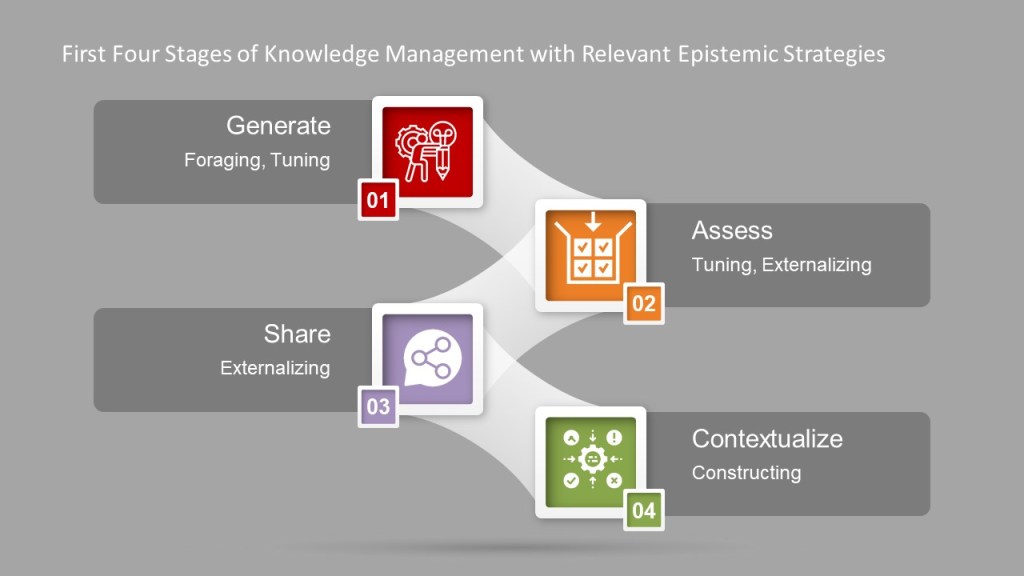
The first four phases of knowledge management are all about identifying and creating meaning and then making that meaning usable. Knowledge management is a set of epistemic actions, creating knowledge through interaction. This interaction is a way of creating a partnership between what happens in the head with everything in the world – Work-as-Imagined and Work-as-Done.
There are really four themes to a set of epistemic actions:
Foraging: Locating resources that will lead to understanding
Tuning: Adjusting resources to align with desired understanding
Externalizing: Moving resources out of the head and into the world
Constructing: Forming new knowledge structures in the world
Four Themes Mapped to Firts 4 Phases of Knowledge Management
Theme
Epistemic Interaction
Means
Foraging
Locating resources that will lead to understanding
Searching
Searching happens when you need information and
believe it exists somewhere.
Searching depends on how we articulate or
information needs.
Probing
“Tell me more.” Probing happens when the information
you have isn’t quite enough. You are probing when you take the next step,
move to the next level, and obtain more salient specifics. Probing is about
drilling down and saying “show, explain, and reveal more about this.”
We can probe to reveal new patterns, structures and
relationships. It brings to light new information that helps us to reconsider
what we already know.
Animating
Animating is when we initiate and control motion in
an information source. It includes learning-by-doing.
Collecting
Collecting is how we gather foraged information and
tuck it away for future use.
Tuning
Adjusting resources to align with desired
understanding
Collecting
Cloning
Cloning lets us take information from one situation
and use it in another.
Cutting
Cutting is the way we say “this matters”, that “I
need this part, but not the rest.”
Filtering
Filtering reduces complexity by reducing clutter to
expose salient details.
Externalizing
Moving resources out of the head and into the world
Annotating
Annotating is how we add context to information. How
we adapt and modify the information to the needed context.
Linking
Connecting bits of information together. Forming
conceptual maps.
Generating
Introducing new knowledge into the world.
Chunking
Grouping idenpendent yet related information together.
Constructing
Forming new knowledge structures in the world
Chunking
Composing
Producing a new, separate structure from the
information that has its own meaning and purpose.
Fragmenting
Taking information and breaking it apart into usable
components.
Rearranging
The art of creating meaningful order.
Repicturing
Changing the way the information is represented to
create understanding.
The format is pivotal. The difficulties we have in quality are really not much different from elsewhere in society in that we are surrounded by confusing documentation and poorly presented explanations everywhere we look, that provide information but not understanding. Oftentimes we rely on canards of “this is what is expected,” “this is what works” – but rarely is that based on anything more than anecdotal. And as the high incidence of issues and the high cost of training shows, less than adequate.
There is a huge body-of-knowledge out there on cognitive-friendly design of visuals, including documentation. This is an area we as a quality profession need to get comfortable with. Most important, we need to give ourselves permission to adapt, modify and transform the information we need into a shape that aids understanding and makes everyone a better thinker.
Work-as-Prescribed (and work-as-instructed) is the creation of tools and technologies to help us think better, understand more and perform at our peak.
Locus of Understanding
Looking at the process at the right level is key. Think of Work-as-Prescribed as a lens. Sometimes you need a high-powered lens so that you can zoom in on a single task. Other times, you need to zoom out to see a set of tasks, a whole process, or how systems interact.
This is the locus of understanding, where understanding happens. When we take this position, we see how understanding is created. Adopting the locus of understanding means going to the right level for the problem at hand. When we apply it to Work-as-Prescribed we are applying the same principles as we do in problem-solving to developing the right tools to govern the work.
An important way to look is distributed cognitive resources, which means anything that contributes to the cognitive work being done. Adjusting the locus of understanding means that you can, and should, treat an SOP as a cognitive resource. Some of the memory is in your head and some is in the SOP. Work-as-prescribed is a cognitive resource that we distribute, routinely and casually across the brain and our quality system in the form of documents and other execution aids.
Other tools, like my favorite whiteboard, also serve as distributed cognitive resources.
So, as our documents and other tools are distributed cognitive resources it behooves us to ensure they are based on the best cognitive principles possible to drive the most benefit.
As an aside, there is a whole line of thought about why some physical objects are better at distributed cognitive resources than electronic. Movement actually matters.
Taking it even further (shifting the locus) we can see the entire quality system as a part of a single distributed cognitive system where cognitive work is performed via the cognitive functions of communicating, deciding, planning, and problem-solving. These cognitive functions are supported by cognitive processes such as perceiving, analyzing, exchanging, and manipulating.
Cognitive Activity in Work-As-Prescribed
The tools we develop to provide distributed cognitive activity strive to:
Provide short-term or long-term memory aids so that memory load can be reduced.
Provide information that can be directly perceived and used such that little effort is needed to interpret and formulate the information explicitly.
Provide knowledge and skills that are unavailable from internal representations.
Support perceptual operators that can recognize features easily and make inferences directly.
Anchor and structure cognitive behavior without conscious awareness.
Change the nature of a task by generating more efficient action sequences.
Stop time and support perceptual rehearsal to make invisible and transient information visible and sustainable.
Aid processibility by limiting abstraction.
Determine decision making strategies through accuracy maximization and effort minimization.
Driving Work-As Prescribed
As we build our requirements documents, our process and procedure, there are a few principles to keep in mind to better tap into distributed cognitive resources.
Plan for the flow of information: Think about paths, relationships, seams, edges and other hand-offs. Focus on the flow of information. Remember that we learn in a spiral, and the content needed for a novice is different from that of an expert and build our documents and the information flow accordingly. This principle is called Sequencing.
Break information down into pieces: Called, Chunking, the grouping together of information into ideally sized pieces. When building Work-As-Prescribed pay close attention to which of these chunks are reusable and build accordingly.
The deeply about context: How a tool is used drives what the tool should be.
Think deeply about information structures: Not all information is the same, not every example of Work-as-Prescribed should have the same structure.
Be conscientiousabout the digital and physical divide: Look for opportunities to integrate or connect these two worlds. Be honest of how enmeshed they are at any point in the system.
We are building our Work-as-Prescribed through leveraging our quality culture, our framework for coordinating work. Pay attention to:
Shared Standards – Ways we communicate
Invisible Environments – Ways we align, conceptually
When design process, procedure and task documentation leverage this principles by build blocks, or microcontent, that is:
about one primary idea, fact, or concept
easily scannable
labeled for clear identification and meaning, and
appropriately written and formatted for use anywhere and any time it is needed.
There is a common miscomprehension that simple means short. That just isn’t true. Simple means that it passes a test for the appropriateness of the size of a piece of content of providing sufficient details to answer a specific question for the targeted audience. The size of the content must effectively serve its intended purpose with efficiency, stripping off any unnecessary components.
We need to strive to apply cognitive thinking principles to our practice. The day of judging a requirements document by its page length is long over.
Constituents of cognitive thinking applied to Work-As-Prescribed
Risk can be associated with a number of different types of consequences, impacting different objectives. The types of consequences to be analyzed are decided when planning the assessment. The context statement is checked to ensure that the consequences to be analyzed align with the purpose of the assessment and the decisions to be made. This can be revisited during the assessment as more is learned.
Methods used in analyzing risks can be qualitative, semiquantitative, or quantitative. The decision here will be on the intended use, the availability of reliable data, and the decision-making needs of the organization. In ICH Q9 this is also the level of formality.
Risk
Is….
The combination of the probability of the occurrence of the harm
and the severity of that harm.
The effect of uncertainty on objectives
Often characterized by reference to the potential event and
consequences or combination of these
Often expressed in terms of a combination of the consequences of
an event (including in changes in circumstances) and the associated
likelihood of the occurrence
Qualitative assessments define consequence (or severity), likelihood, and level of risk by significance levels, such as “high,” “medium,” or “low.” They work best when supporting analysis that have a narrow application or are within another quality system, such as change control.
Qualitative
Below is a good way to break down consequences and likelihood for a less formal assessment.
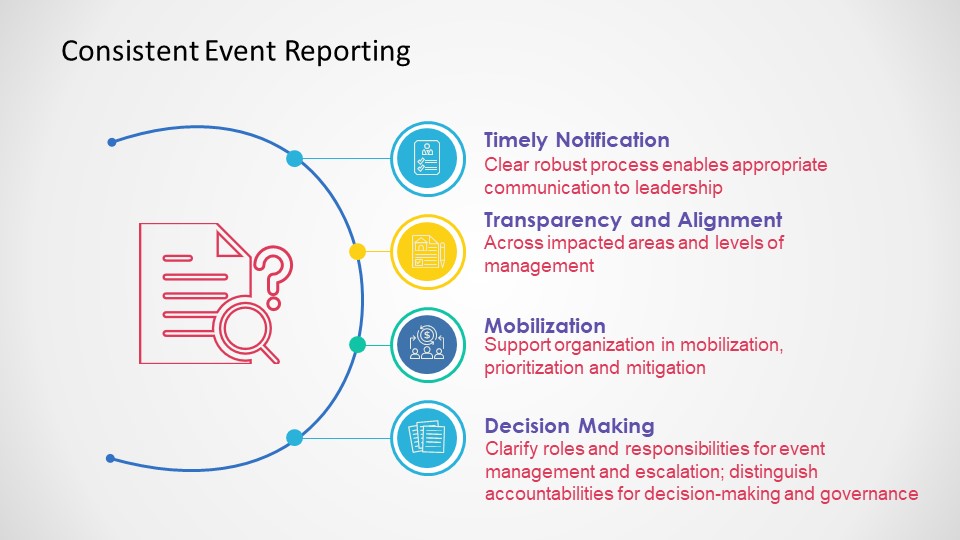
Event management systems need to have an escalation mechanism to ensure critical events are quickly elevated to a senior level to ensure organization-wide timely reactions.
Consistent Event Reporting
There are many reasons for a fast escalation.
Events that trigger reporting to Regulatory Agencies (e.g. Serious Breach, Urgent Safety Measures (UK), Field Alerts, Biological Product Deviation, Medical Device Report)
Events that require immediate action to prevent additional harm from across the organization
Events that require marshalling resources from large parts of the organization
•Reference GxP area for Impact
resulting from/linked to system error/failure
•Product Quality/ CMC events in
accordance with MRB criteria (or other events of similar scope of impact)
•Impact to study integrity
•Impact to subject’s safety, rights or
welfare
•Gaps in reporting/ collection of
potential AEs
•Impact to study integrity
•Impact to study integrity
•System design, testing, deployment,
upgrade, etc. event impacting GxP data integrity or regulatory compliance
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Potential Falsified or Counterfeit
Product
•Potential Fraud or Misconduct
•Potential Fraud or Misconduct
•Credible Risk of Product Shortage
•Quality event with patient safety
risk/gap
•GxP Data Breach
•Potential Product Recall
•Significant Quality Event Notified to
Regulatory Authority
•System error or failure with
significant GxP compliance impact
·Potential Critical Finding Resulting from
Regulatory Authority Inspection or Audit by External Body/Third Party
·Quality Event/Observation Classified
as Critical (Event or Internal Audit) Notification from Regulatory Authority
or other External Authority of Findings of Significant/Critical Quality
Deficiency (inspection or other than through inspection)
oe.g.; Refusal to File, Notification
of Inadequate Response to Inspection Findings (e.g.; Other Action Indicated
(FDA classification), Warning Letter
You can drill down to a lower, more practical level, like this
Escalation Criteria
Examples of Quality Events for Escalation
Potential to adversely affect
quality, safety, efficacy, performance or compliance of product (commercial
or clinical)
•Contamination (product, raw material,
equipment, micro; environmental)
•Product defect/deviation from process
parameters or specification (on file with agencies)
•Significant GMP deviations
•Incorrect/deficient labeling
•Product complaints (significant PC,
trends in PCs)
•OOS/OOT (e.g., stability)
Product counterfeiting, tampering, theft
•Product counterfeiting, tampering, theft reportable to Health
Authority (HA)
•Lost/stolen IMP
•Fraud or misconduct associated with counterfeiting, tampering,
theft
•Potential to impact product supply (e.g., removal, correction,
recall)
Product shortage likely to
disrupt patient care and/or reportable to HA
•Disruption of product supply due to
product quality events, natural disasters (business continuity disruption),
OOS impact, capacity constraints
Potential to cause patient harm associated with a product
quality event
•Urgent Safety Measure, Serious Breach, Significant Product
Compliant, Safety Signal that are determined associated with a product
quality event
Significant GMP
non-compliance/event
•Non-compliance or non-conformance
event with potential to impact product performance meeting specification,
safety efficacy or regulatory requirements
Regulatory Compliance Event
•Significant (critical, repeat) regulatory inspection findings,
lack of commitment adherence
•Notification of directed/for cause inspection
•Notification of HA correspondence indicating potential
regulatory action
An updated and expanded version of this is found here.
Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
Regulation/Guidance
States
ICH E-6 (R2) Section 4.5.1-4.5.4
4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion.
ICH E3, section 9.6
The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report
21CFR 312.53(vi) (a)
investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.”
21CFR 56.108(a)
IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects.
21 CFR 56.108(b)
“IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.”
45 CFR 46.103(b)(5)
Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB.
FDA Form-1572 (Section 9)
lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.”
A few key regulations and guidances (not meant to be a comprehensive list)
How Protocol Deviations are Implemented
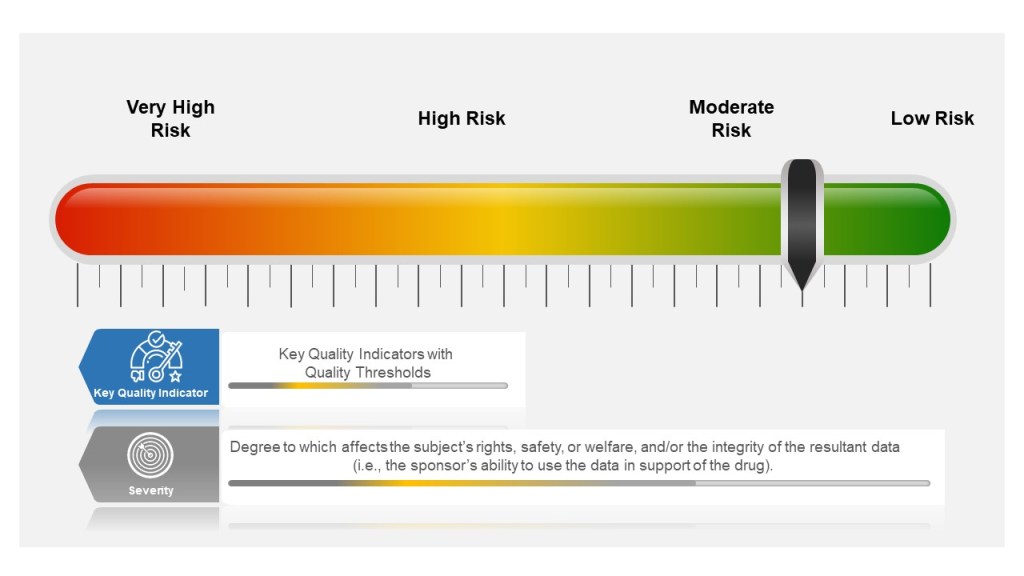
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
Events happen beyond the Protocol. These need to be managed appropriately as well.
The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Event Classification for Clinical Protocols and GCPs
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
Events Happen
Consistent Event Reporting is Critical
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are: